
Abstract
Recently, new complexities in cell fate decision for helper T cells have emerged. One new lineage, which has come to be called Th17 cells, selectively produces proinflammatory cytokines including interleukin-17 (IL-17, A and F), IL-21, and IL-22. In conjunction with transforming growth factor β-1 (TGFβ-1), IL-6, IL-21, and IL-23, which activate the transcription factor, signal transducer, and activator of transcription 3 (Stat3), the expression of another transcription factor, retinoic acid-related orphan receptor-γt (RORγt) leads to the differentiation of Th17 cells in mice. Other cytokines including IL-2, IL-4, interferon-γ (IFN-γ), and IL-27 inhibit Th17 differentiation. However, IL-2 acting with TGFβ-1 induces differentiation of naïve CD4+ T cells to become regulatory T cells (Tregs). Th17 cells are now known to play an important role not only in the pathogenesis of inflammation and autoimmune diseases, but also host defense against extracellular bacteria. Conversely, extensive data substantiate the role of Tregs as essential in maintenance of peripheral tolerance. Selectively targeting Tregs and Th17 cells are likely to be important strategies in the treatment of inflammatory and autoimmune diseases in humans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is well-established that the innate immune response leads to activation of adaptive immunity, shaping the proper response, depending upon the pathogen eliciting the reaction. Conversely, though, it is also clear that in adaptive immune response, especially, CD4+ or helper T cells orchestrate responses to effectively eliminate pathogens. Classically, helper T cells were considered to differentiate into two lineages, T helper 1 (Th1) cells or Th2 cells [1]. In addition to ligation of the T-cell receptor (TCR) and co-stimulatory receptors, a suitable cytokine milieu is required for fate decision of helper T cells [1]. In particular, interleukin-12 (IL-12), which is produced by activated antigen-presenting cells (APCs), promotes Th1 differentiation [2], whereas IL-4, produced by activated T cells and some innate immune cells, drives Th2 differentiation [1].
While the Th1/Th2 paradigm provided invaluable insights for immunologists, its limitations were also clear. What was the most obvious was the inability of this dichotomy to explain autoimmune diseases. More recent studies have shown the existence of the additional subpopulations of T helper cells that are also important in immunoregulation and host defense. One important subset is CD4+CD25+, regulatory T cells (Tregs) [3, 4]. Tregs suppress the proliferation and function of effector T cells, and attenuate immune responses against self or nonself-antigens [3, 4].
Another recently identified new fate of T cells is Th17 cells that selectively produce IL-17 (A and F) [5, 6]. In the past three years, discoveries pertaining to this newly identified T helper subset in humans and mice have accumulated with tremendous speed [7–10]. It is also clear that the notion of a single type of Th17 cell may not be correct, as there seems to be further complexity in terms of the cytokines produced by these cells. In this review, we will discuss the historical developments that led to a new understanding of the role of T cells in autoimmune diseases and recent advances that have led to even newer insights into T cell biology.
Pathogenicity of IL-23, but not IL-12, in autoimmune diseases
Until a few years ago we thought we knew which subset of helper T cells was responsible for host defense or exacerbation of autoimmune diseases and we thought that answer fit well with the standard Th1/Th2 cells paradigm [11]. Th1 cells produce interferon-γ (IFN-γ) and lymphotoxin, and are responsible for protection against intracellular pathogens such as viruses, mycobacteria, and protozoa [11]. Produced by activated APCs, IL-12 activates the transcription factor, signal transducer, and activator of transcription 4 (Stat4) [2]. Stat4-dependent signaling in conjunction with TCR-dependent signals induces the expression of T-box-expressed-in-T-cells (T-bet), the so-called master regulator of Th1 differentiation [2]. Recently, in humans, it has been shown that STAT4 polymorphisms are associated with rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) [12]. IFN-γ also activates Stat1 which further induces T-bet to form an autocrine feedback loop for IFN-γ production [2]. T-bet promotes not only IFN-γ production but also suppression of Th2 cytokines production by T cells [2]. On the other hand, Th2 cells produce IL-4, IL-5, and IL-13 [1]. IL-4 activates Stat6, which up-regulates expression of GATA-binding protein 3 (GATA-3) [1]. Th2 cytokines are potent activators of B-cell Immunoglobulin E production, eosinophil recruitment, and mucosal expulsion mechanisms, and are essential for promoting host defense against helminths and other parasites [1]. In addition, Th2 cells have been shown to mediate allergic diseases such as asthma, rhinitis, and atopic dermatitis [11].
Traditionally, autoimmune diseases had been assumed to be associated with dysregulated Th1 responses [13]. Because treatment with anti-IL-12p40 antibody was effective in Crohn’s disease (CD) and psoriasis [14, 15], it was further assumed that IL-12-mediated IFN-γ production and Th1 response were involved in the pathogenesis of autoimmunity. However, it was shown that IFN-γ deficiency exacerbated rather than ameliorated mouse models of autoimmune diseases such as experimental autoimmune encephalomyelitis (EAE) [16]. Subsequently, a new cytokine, which consists of IL-23p19 and IL-12p40, was identified and termed IL-23 [17]. Interestingly, the IL-23 receptor (IL-23R) forms dimers with the IL-12Rβ1 chain shared by IL-12 and IL-23 [18]. IL-23R is predominantly expressed on T and natural killer (NK) cells [18, 19]. IL-23 activates Janus kinase 2 (Jak2) and tyrosine kinase 2 (Tyk2), which in turn leads to activation of Stat1, Stat3, Stat4, and Stat5, among which Stat3 is the predominant factor phosphorylated by this cytokine [17, 18].
Of note, IL-23p19-deficient mice or IL-12p40-deficient (IL-12/IL-23-deficient) mice were resistant to collagen-induced arthritis (CIA) and EAE [20, 21]. However, IL-12p35-deficient mice had increased susceptibility to CIA and EAE [20, 21]. These studies directly negated the pathogenic role of the IL-12/IFN-γ axis in autoimmunity. This was an important piece of information that led to the unraveling of the Th1/Th2 paradigm as an explanation of autoimmunity. Accordingly, additional studies on IL-23p19-deficient mice and the anti-IL-23p19 antibody revealed that IL-23, but not IL-12, was the culprit in autoimmunity, at least in mouse disease models [21–23]. The pathogenic role of IL-23 was also confirmed by the phenotype of IL-23p19 transgenic mice, in which premature death, systemic inflammation, anemia, and elevated serum levels of inflammatory cytokines and acute phase proteins were observed [22]. In human disease, the mRNA levels of both IL-23p19 and IL-12p40 were shown to be increased in skin lesions of psoriatic patients [24], suggesting that elevated IL-23 contributed to the pathogenesis of psoriasis. Therefore, IL-23 began to attract attention as a factor crucial to inflammation and autoimmune diseases.
The IL-23/IL17 axis in inflammation
While it was becoming clear that IL-23 was the culprit in autoimmune and inflammatory diseases, the question remained, how was it acting? Another inflammatory cytokine, IL-17, was noted to be produced by activated purified T cells in the presence of LPS-stimulated dendritic cells (DCs) [5]. It was also found that IL-23 promoted IL-17 production from memory T cells and this production was suppressed by treatment with anti-IL-12p40 antibody [5].
Now IL-17 is recognized as the founding member of a family of proinflammatory cytokines: IL-17 (IL-17A), IL-17B, IL-17C, IL-17D, IL-17E (known as IL-25), and IL-17F [6]. IL-17A and IL-17F induce the production of various proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), IL-1β, and IL-6, and CXC chemokines from monocytes, airway epithelial cells, vein endothelial cells, and fibroblasts to mount defense against extracellular bacteria such as Klebsiella pneumonia, Bacteroides fragilis, and Mycobacterium tuberculosis [6, 25, 26]. These responses result in the recruitment, activation and migration of neutrophils to the sites of inflammation and infection [6]. CD4+ T cells are major producers of IL-17, but in addition CD8+ T cells, neutrophils, γδ T cells, and invariant natural killer T (NKT) cells also produce this cytokine [26–28].
Various reports associated IL-23 with IL-17 production with the onset or exacerbation of inflammation [20–23, 29]. For instance, IL-23p19-deficient mice showed reduced incidence of CIA, with fewer IL-17-producing CD4+ T cells [20]. Consistent with this report, treatment with anti-IL-23p19 antibody reduced serum levels of IL-17 and inhibited development of EAE [29]. Moreover, IL-23 was found to be essential for production of IL-17 and IL-6 in colitis mouse models [22, 23]. Regardless of exactly how IL-23 works, these reports supported the idea that an IL-23/IL-17 axis, but not an IL-12/IFN-γ axis, was a crucial pathway in the pathogenesis of various autoimmune diseases [21, 30].
Regulation of Th17 differentiation
Although many results linked IL-23 with IL-17 production in CD4+ T cells, less was known about how Th17 cells arose from naïve CD4+ T cells. It was very clear that development of IL-17-producing T cells was not dependent on the cytokines and the transcription factors required for Th1 or Th2 differentiation and then it was proposed that IL-17-producing CD4+ T cells represented a new subset of helper T cells (Th17 cells), which were crucial in mediating inflammatory responses [30–32]. While there was evidence that IL-23 stimulated CD4+ T cells to express various genes, including IL-17, and that IL-23-stimulated T cells were important in the pathogenesis of EAE [30], it was less clear whether IL-23 was the factor that drove naïve CD4+ T cells to become IL-17 producers.
It was especially puzzling that naïve CD4+ T cells did not express IL-23R [18]. In a search for optimum culture conditions for Th17 differentiation it was recognized that naïve CD4+ T cells cultured with activated DCs produced IL-17 [16]. It was then recognized that the combination of IL-6 and transforming growth factor β-1 (TGFβ-1), with appropriate stimulation of TCR, were the optimum stimuli inducing Th17 differentiation (Fig. 1a) [16]. Accordingly, TGFβ-1-deficient mice showed impaired Th17 development, whereas addition of TGFβ-1 rescued the number of Th17 cells [7, 33]. Thus it became clear that IL-23 was not the direct initiator of Th17 production [16], but rather was an important factor for expanding and maintaining Th17 cells in vivo [34].

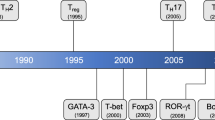
New T cell lineages in mice (a) and humans (c). When stimulated by APCs such as DCs and macrophages, and cognate peptide, naïve helper T cells differentiate into lineages determined by the cytokine milieu. TGFβ-1 is a critical factor in the differentiation of new two lineages in mice T cells. In conjunction with TCR stimulation, the combination of TGFβ-1 and IL-2 (and other γc cytokines) induces the expression of Foxp3, leading to the differentiation of naïve CD4+ T cells into anti-inflammatory Tregs (a). However, in the presence of TGFβ-1 with IL-6 or IL-21, which activates Stat3 and up-regulates RORγt, naïve CD4+ T cells develop to become pro-inflammatory Th17 cells (a). Th17 differentiation is suppressed by IFN-γ, IL-4, IL-2, IL-25, IL-27, and retinoic acid in mice (b). Both Th17 cells and Tregs are also present in humans (c). TGFβ-1, IL-1β, and IL-2 in combination with IL-6, IL-21 or IL-23 are necessary for human Th17 differentiation (c). APCs antigen-presenting cells; iTregs inducible Tregs
Autocrine regulation of Th17 differentiation by IL-21
In the classic Th1/Th2 paradigm, T cell subsets produce factors that promote their differentiation and constrain the differentiation of the opposing lineage. It was of interest in this regard that Th17 cells were found to selectively produce IL-21 [35–38]. IL-21 has structural homology with IL-2 and IL-15, and the initial report showed the effects of IL-21 produced by CD4+ T cells and NKT cells on the proliferation and function of NK cells, B cells, and T cells [39, 40]. Recently though, it has also been found that the autocrine production of IL-21 by Th17 cells promotes Th17 differentiation while inhibiting IFN-γ production [35–38]. Accordingly, the deficiency of IL-21 (or its receptor), or blocking of IL-21, attenuates the generation of Th17 cells [35–37].
Transcription factors for Th17 differentiation
To precisely understand the molecular mechanisms of Th17 differentiation, it is important to define the transcription factors that directly regulate IL-17 production. All of IL-6, IL-21, and IL-23 activate the transcription factor Stat3 in T cells [38, 41, 42]; accordingly, Th17 differentiation is abrogated in Stat3-deficient T cells [38, 42–45]. Moreover, conditional deletion of Stat3 in T cells also abrogates the development of autoimmune diseases such as EAE and autoimmune pneumonitis in mice [43]. The role of Stat3 in Th17 differentiation seems remarkably direct—i.e. Stat3 binds to the Il17 and Il21 loci, as detected by using chromatin immunoprecipitation (ChiP) assays [38, 41]. In addition, IL-6 and IL-23 promote IL-23R expression and this also is Stat3-dependent [10, 36, 37]. Recently, the importance of Stat3 in human Th17 differentiation has been documented [46].
Like other T cell subsets, Th17 cells also have a lineage-specific transcription factor, namely the retinoic acid-related orphan receptor-γt (RORγt) [8]. Interestingly, RORγt-deficient T cells produce fewer Th17 cells, whereas overexpression of RORγt in T cells promotes IL-17 expression [8]. Furthermore, RORγt-deficient mice are less susceptible to EAE, suggesting that RORγt is a key regulator of Th17 differentiation [8]. Recent studies have shown that RORγt expression is significantly reduced in Stat3-deficient T cells [43–45]. However, the precise molecular mechanisms of Stat3-mediated expression of RORγt are still unclear. Even so, RORγt could be a good candidate to target in treating inflammatory diseases (discussed in further detail below). Also, it has been shown that expression of another retinoic acid related nuclear receptor, RORα also induces Th17 differentiation [47]. RORα seems to synergize with RORγt to promote development of Th17 lineage [47].
Counter-regulation of IL-17 and the Yin/Yang of Th17 cells and Tregs
Because of their potential for inducing immune-mediated damage to the host, it is perhaps not surprising that multiple mechanisms exist to inhibit the production of Th17 cells (Fig. 1b). Indeed, it has been recognized that the Th1-related cytokine IFN-γ inhibits Th17 differentiation [31, 32]. Consistent with this concept, T-bet-deficient mice show enhanced Th17 differentiation [48]. Similarly, the Th2-related IL-4 is also shown to down-regulate Th17 differentiation [49]. However, how IFN-γ and IL-4 inhibit IL-17 production is not yet known. Interestingly, IL-27, an IL-12-related heterodimeric cytokine, also has anti-inflammatory effect by inhibiting the generation of Th17 lineage and leads to limitation of EAE [50]. Like IFN-γ, IL-27 suppresses Th17 differentiation through a STAT1-dependent manner [51, 52].
IL-25 (IL-17E) was also shown to inhibit IL-17 production by promoting Th2 differentiation [53]. These reports suggest a cross-regulatory relationship among Th1, Th2, and Th17 differentiation. However, it still remains to be determined how Th1-related and Th2-related transcription factors negatively regulate IL-17 production.
Another new subset of helper T cells that provides considerable insight into the pathogenesis of autoimmunity is the CD4+CD25+ regulatory T cells (Tregs) [3, 4, 54]. Tregs suppress proliferation of effector T cells and maintain self tolerance by down-regulating immune responses against self or nonself-antigens [3, 4, 54]. The mechanism by which Tregs preserve peripheral tolerance is still not entirely clear; however, they preferentially express cytotoxic T-lymphocyte antigen 4 (CTLA-4), and immunosuppressive cytokines including TGFβ-1 and IL-10 [55–57]. Naturally arising Tregs (nTregs) are generated in the thymus, whereas inducible Tregs (iTregs) arise from naive T cells after antigen exposure and cytokine stimulation in the periphery, especially in the gut [58]. Recently, it has been shown that CD4+CD25+ T cells express the transcription factor, forkhead box protein 3 (Foxp3), which is necessary and sufficient for Tregs to develop and function [59, 60].
In mice, IL-2 and TGFβ-1 were able to differentiate CD4+CD25− naïve T cells into CD4+ CD25+ Foxp3+ Tregs [61, 62]. Therefore, the importance of TGFβ-1 for both Tregs and Th17 lineage led to the speculation that these lineages were related. This putative relationship also led to investigation of the importance of IL-2 in Th17 differentiation [45]. In fact, IL-2 was found to inhibit the expression of RORγt while the enhancing expression of Foxp3 [45]. Furthermore, blockade of IL-2 or deletion of Stat5 promoted Th17 differentiation, suggesting that IL-2 inhibited Th17 lineage by affecting the RORγt–Foxp3 balance [45]. Therefore, helper T cells seem to have a unique way of achieving a balance between Tregs and Th17 cells during ongoing immune responses. Recently, it has been reported that TGFβ-1 can promote either Th17 or Tregs lineage differentiation, depending on its local concentration [63]. Interestingly, Foxp3 may inhibit RORγt activity on its target genes, at least in part, through direct interaction with RORγt [63].
Further complexities of Th17 cells
The roles of Th17 cells are complicated by the fact that these cells also have the ability to produce anti-inflammatory cytokines such as IL-10 [34]. This cytokine has anti-inflammatory properties that suppress functions of DCs and macrophages [64]. IL-10 can be produced by many cell types such as B cells, macrophages, DCs, Th2 cells, and distinct populations of Tregs [64]. Recently, it has been reported that de-novo generation of Th17 cells in the presence of IL-6 and TGFβ-1 leads to the production of IL-10 with up-regulation of RORγt and IL-17 [34]. Furthermore, IL-1β, but not IL-23, potentiates IL-10 production in Th17 cells along with IL-6 and TGFβ-1 [34]. Paradoxically, IL-6 and TGFβ-1 seem to drive initial Th17 lineage commitment but also restrain the pathogenic potential of Th17 cells by producing IL-10 also [34].
Another cytokine produced by Th17 cells is IL-22 [25, 65, 66]. IL-22 belongs to the IL-10 family of cytokines which include IL-19, IL-20, IL-24, and IL-26. Expressed in activated T cells, Th1 cells and NK cells [67], IL-22 induces expression of various antimicrobial peptides such as β-defensins in skin, respiratory, and digestive tissues, which express IL-22R [68]. In fact, expression of IL-22 and β-defensins is higher in skin from patients with psoriasis and atopic dermatitis than in that from healthy individuals [69].
While it is clear that IL-22 and IL-17 can contribute to host defense and to pathogenesis of various autoimmune diseases [25], it is also notable that IL-22 also has important anti-inflammatory effects [70]. Specifically, IL-22-expressing Th17 cells provided protection in models of hepatitis [70]. Recently, IL-22 also has been shown not to be required for the development of EAE in mice [71]. These facts indicate the complexity of Th17 cells, which produce multiple cytokines and have diverse functions during inflammation. As such, their relevance may vary among different autoimmune diseases.
Human Th17 differentiation
While a large body of evidence has documented that the best recipe for generating Th17 cells from murine naïve CD4+ T cells is the combination of TGFβ-1 and IL-6 or IL-21, several groups initially reported that the regulation of human Th17 differentiation was quite distinct (Fig. 1c) [9, 10, 66, 72–74]. Specifically, TGFβ-1 was initially considered to have inhibitory effect on human IL-17 production; however, the role of TGFβ-1 in human Th17 differentiation has recently become clearer. While TGFβ-1 alone had an inhibitory effect on production of IL-17 by T cells, culturing human naïve CD4+ T cells from cord blood in serum-free medium revealed a requirement of TGFβ-1 for Th17 differentiation [72]. TGFβ-1 seems to have dual effects on human Th17 differentiation in a dose-dependent manner. While TGFβ-1 is required for the expression of RORγt, in human naïve CD4+ T cells from cord blood, TGFβ-1 can inhibit the function of RORγt at high doses [72]. However, inflammatory cytokines (IL-1β, IL-6, and IL-21 or IL-23) relieve the latter effect of TGFβ-1 [72]. By using serum-free medium it has thus been clarified that the optimum conditions for human Th17 differentiation are TGFβ-1, IL-1β, and IL-2 in combination with IL-6, IL-21 or IL-23 [73–75]. Of these latter cytokines, IL-23 seems the most effective [10, 66, 72, 73]. A key aspect of Th17 regulation is the regulation of IL-23R [10]. Naïve CD4+ T cells express low levels of this receptor, but the combination of TGFβ-1, IL-1β, and IL-23 can strongly induce this receptor [10, 72]. Thus, IL-23 promotes expression of its own receptor, allowing it to be an effective inducer of IL-17.
It is of note is that CCR6+IL-23R+ human memory CD4+ T cells are major producers of IL-17 [75, 76]. Because CCR6+CD4+ T cells, but not CCR6-CD4+ T cells, secrete IL-17 and expression of IL-23R, CCR6, and RORγt is up-regulated during Th17 differentiation [72], these factors seem to be essential for development of human Th17 lineage. Importantly, it has been shown that mutations presumed to underlie hyper-IgE syndrome (HIES, “Job's syndrome”) are identified in the STAT3 gene [46]. Purified naïve CD4+ T cells from HIES individuals cannot differentiate into Th17 cells in vitro and have lower expression of RORγt [46], suggesting that Stat3 signaling is also a crucial factor for the generation of human Th17 cells.
Th17 cells in autoimmune diseases
Although Th17 cells are important and essential subsets participating in host defense against extracellular antigens, evidence implicating Th17 cells as a causal factor of autoimmune diseases is also mounting [25]. Following extensive analysis of mouse disease models such as EAE, CIA, and IBD, it has also become evident that IL-17 is a contributing factor in human autoimmune diseases. For instance, perivascular IL-17-producing T cells are present in brain lesions of active MS patients and these cells are reduced in quiescent MS patients [77]. Moreover, DCs from MS patients secrete more IL-23 than healthy controls, and T cells from MS patients secrete more IL-17 than healthy controls [78]. Also, elevated levels of IL-17 have been noted in the sera and in tissues of patients with RA, IBD, and psoriasis [25]. We will see more evidence confirming the roles of IL-17 in human diseases and inflammation in the future.
Recent genetic data has linked polymorphisms of the Il23r gene to various human diseases such as ankylosing spondylitis, CD, and psoriasis [79–81]. Accordingly treating CD and psoriasis patients with anti-IL-12p40 antibody has resulted in some success [14, 15]. These results argue strongly for a role of the IL-23/IL-17 axis in human autoimmune diseases.
Th17 cells in rheumatoid arthritis
It is of particular interest to evaluate the contribution of Th17 cells to the pathogenesis of RA, because targeting pathogenetic Th17 might be a reasonable strategy for treating RA. First, IL-17-deficient mice fail to develop CIA when challenged [82]. Consistent with this, treatment with neutralizing IL-17 monoclonal antibody ameliorates joint inflammation in CIA models [83]. Furthermore, it has been reported that active immunization using virus-like particles conjugated with recombinant IL-17 leads to high levels of anti-IL-17 antibodies in mice and reduces scores of disease severity in CIA [84]. These results highlight the role of IL-17 as an exacerbation factor in rodent arthritis models.
Because expression of IL-17 and IL-23p19 was observed in the synovial fluid and serum of RA patients, these factors were presumed to be pathogenic [85]. In particular, IL-17 up-regulates the production of IL-1β and TNF-α from APCs in arthritic joints [86, 87]. Moreover, IL-17 induces IL-6, IL-8, and IL-23 from RA synovial fibroblasts and chemokines such as CCL20/MIP3α from synoviocytes to induce migration of T cells and DCs into inflammatory lesions [86, 87]. IL-17 also promotes cartilage breakdown by inducing matrix metalloproteinases-1 (MMP-1) from RA synovium [86, 87]. Additionally, IL-17 induces cyclooxygenase-2 (COX-2)-dependent prostaglandin E2 (PGE2) synthesis by osteoblasts and it then induces expression of receptor activator of NF-κB ligand (RANKL) to differentiate osteoclast progenitors into mature osteoclasts [86, 87]. Pro-oxidants such as nitric oxide are also produced by chondrocytes and osteoblasts in response to IL-17 [86, 87]. Taken together, these multiple factors induced by IL-17 seem to promote bone resorption, extracellular matrix degradation, synovium proliferation, angiogenesis, and recruitment and activation of immune cells for bone erosion and articular destruction in RA joints. This being said, there is little evidence of T cell proliferation in RA and we still do not know for certain the cellular source of IL-17 in RA synovium [88].
For these reasons, IL-17 seems to be a rational target in the treatment of RA, and Phase I/II clinical trials of anti-IL-17 monoclonal antibody (AIN457) have commenced (http://www.clinicaltrials.gov/). It has been reported that combining inhibitors to block TNF-α and IL-17 has the synergistic effect of suppressing IL-6 production and collagen degradation in the synovium and bone of RA patients [89]. Therefore, anti-IL-17 therapy may become an alternative for treating patients unresponsive to conventional anti-TNF therapy. It still remains to be seen whether blocking IL-17 will affect other proinflammatory cytokines for example IL-6, IL-1, and IL-22 in RA patients. More studies will shed light on the validity of anti-IL-17 therapy.
While the role of IL-6 in regulating IL-17 production has only recently been appreciated, the role of IL-6 as a proinflammatory cytokine has long been recognized. For this reason, targeting IL-6 in RA has been considered for some time. Indeed the efficacy of humanized anti IL-6R monoclonal antibodies (tocilizumab/atlizumab) in the treatment of adults and children with arthritis has recently been reported [90]. Also, in April 2008 tocilizumab was approved in Japan for treatment of RA, and Phase III trials have been completed in the US (http://www.clinicaltrials.gov/). To what extent their efficacy relates to inhibition of IL-17 will be important to ascertain. Similarly, in the US, recombinant human anti-IL-1Rα antagonist (anakinra) has been approved for use in the treatment of RA [91]. Recently, more IL-1-targeting therapies for RA patients are in Phase II studies including anti-IL-1β monoclonal antibody (ACZ885), human monoclonal antibody to IL-1R (AMG108), and IL-1 Trap/rilonacept (http://www.clinicaltrials.gov/). Although IL-1 has been reported to contribute to Th17 differentiation in mouse and man, it remains to be determined whether therapeutic targeting of IL-1 will substantially affect IL-17 in RA. On the basis of on the efficacy of anti-IL-12p40 therapy in human autoimmune diseases [14, 15], a humanized monoclonal anti-IL-12p40 antibody (ABT-874) was tested in RA [92]. However, this antibody was not shown to be effective because of the trial design [92]. Another strategy for targeting IL-12/IL-23 is apilimod mesylate (STA-5326), an oral inhibitor of IL-12p35/p40, which has been tested in a Phase II study for RA (http://www.clinicaltrials.gov/). Whether selectively targeting IL-23p19 will be more efficacious or have a better safety profile remains to be determined.
In principle, targeting IL-21 might also be efficacious in treating RA in which IL-17 is involved in immune-mediated damage. Importantly, the common gamma chain and JAK3 play a pivotal role in the signal transduction of IL-21 [25]. In fact, a JAK3 inhibitor (CP-690550) is about to be tested in RA (Phase II trial) (http://www.clinicaltrials.gov/). It will be important to know whether the efficacy of this drug is related to inhibition of IL-17.
Concluding remarks
The past four years have witnessed major revisions of our views on the lineage commitment of helper T cells. The conventional Th1/Th2 dichotomy has been replaced by more complex multi-lineage models which do a much better job of explaining the pathogenesis of autoimmunity (Fig. 1). It now seems that Th17 cells are one of the more important villains in the pathogenesis of autoimmune diseases. However, it is of note that they are also essential for host defense against extracellular bacteria. Therefore, the challenge will be to keep Th17 function in check to control autoimmunity, while maintaining host defense at the same time. In this regard, we still need to learn what are the most critical factors for Th17 differentiation. We also need to better understand which human autoimmune diseases are truly mediated by IL-17 and, by extension, which diseases will benefit most from anti-IL-17 therapy. With the current pace of investigations on Th17, this field has a great potential for advances. There is no doubt that IL-17 and the other proinflammatory cytokines are likely to be relevant therapeutic targets in the treatment of human autoimmune diseases.
References
Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–44.
Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–58.
Shevach EM. Regulatory T cells in autoimmunity. Annu Rev Immunol. 2000;18:423–49.
Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62.
Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–4.
Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–76.
Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–4.
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33.
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–9.
Chen Z, Tato CM, Muul L, Laurence A, O’Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007;56:2936–46.
Paul WE, Seder RA. Lymphocyte responses and cytokines. Cell. 1994;76:241–51.
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357:977–86.
Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol. 1996;156:5–7.
Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, et al. Anti-interleukin-12 antibody for active Crohn’s disease. N Engl J Med. 2004;351:2069–79.
Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, et al. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med. 2007;356:580–92.
Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89.
Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25.
Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–708.
Belladonna ML, Renauld JC, Bianchi R, Vacca C, Fallarino F, Orabona C, et al. IL-23 and IL-12 have overlapping, but distinct, effects on murine dendritic cells. J Immunol. 2002;168:5448–54.
Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–7.
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8.
Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–6.
Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–83.
Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125–30.
Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67.
Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–9.
Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol. 2003;170:2106–12.
Michel ML, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med. 2007;204:995–1001.
Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–26.
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40.
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41.
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8.
McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–7.
Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–7.
Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–3.
Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74.
Wei L, Laurence A, Elias KM, O’Shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282:34605–10.
Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408:57–63.
Coquet JM, Kyparissoudis K, Pellicci DG, Besra G, Berzins SP, Smyth MJ, et al. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J Immunol. 2007;178:2827–34.
Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci USA. 2006;103:8137–42.
Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–63.
Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, et al. Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–7.
Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–81.
Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O’Malley JT, et al. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol. 2007;178:4901–7.
Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–6.
Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39.
Mathur AN, Chang HC, Zisoulis DG, Kapur R, Belladonna ML, Kansas GS, et al. T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood. 2006;108:1595–601.
Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. 2006;18:349–56.
Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–36.
Takeda A, Hamano S, Yamanaka A, Hanada T, Ishibashi T, Mak TW, et al. Cutting edge: role of IL-27/WSX-1 signaling for induction of T-bet through activation of STAT1 during initial Th1 commitment. J Immunol. 2003;170:4886–90.
Kamiya S, Owaki T, Morishima N, Fukai F, Mizuguchi J, Yoshimoto T. An indispensable role for STAT1 in IL-27-induced T-bet expression but not proliferation of naive CD4+ T cells. J Immunol. 2004;173:3871–7.
Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–70.
Thornton AM, Shevach EM. CD4+ CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–96.
Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190:995–1004.
Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, et al. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2005;201:737–46.
Kullberg MC, Hay V, Cheever AW, Mamura M, Sher A, Letterio JJ, et al. TGF-beta1 production by CD4+ CD25+ regulatory T cells is not essential for suppression of intestinal inflammation. Eur J Immunol. 2005;35:2886–95.
Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3:253–7.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6.
Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–41.
Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+ CD25− naive T cells to CD4+ CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86.
Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–6.
Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–40.
Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765.
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–9.
Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–7.
Wolk K, Kunz S, Asadullah K, Sabat R. Cutting edge: immune cells as sources and targets of the IL-10 family members? J Immunol. 2002;168:5397–402.
Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309–23.
Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–54.
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27:647–59.
Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, et al. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. 2007;179:8098–104.
Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–9.
Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–7.
Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008 (in press).
Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–61.
Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–46.
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–55.
Vaknin-Dembinsky A, Balashov K, Weiner HL. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol. 2006;176:7768–74.
Smith RL, Warren RB, Eyre S, Ho P, Ke X, Young HS, et al. Polymorphisms in the IL-12beta and IL-23R genes are associated with psoriasis of early onset in a UK cohort. J Invest Dermatol. 2008;128:1325–7.
Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Association scan of 14, 500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–37.
Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3.
Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–7.
Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–9.
Rohn TA, Jennings GT, Hernandez M, Grest P, Beck M, Zou Y, et al. Vaccination against IL-17 suppresses autoimmune arthritis and encephalomyelitis. Eur J Immunol. 2006;36:2857–67.
Kim HR, Kim HS, Park MK, Cho ML, Lee SH, Kim HY. The clinical role of IL-23p19 in patients with rheumatoid arthritis. Scand J Rheumatol. 2007;36:259–64.
Miossec P. Interleukin-17 in fashion, at last: ten years after its description, its cellular source has been identified. Arthritis Rheum. 2007;56:2111–5.
Lubberts E. IL-17/Th17 targeting: on the road to prevent chronic destructive arthritis? Cytokine. 2008;41:84–91.
Toh ML, Miossec P. The role of T cells in rheumatoid arthritis: new subsets and new targets. Curr Opin Rheumatol. 2007;19:284–8.
Chabaud M, Miossec P. The combination of tumor necrosis factor alpha blockade with interleukin-1 and interleukin-17 blockade is more effective for controlling synovial inflammation and bone resorption in an ex vivo model. Arthritis Rheum. 2001;44:1293–303.
Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–97.
Cohen SB. The use of anakinra, an interleukin-1 receptor antagonist, in the treatment of rheumatoid arthritis. Rheum Dis Clin North Am. 2004;30:365–80, vii.
Voulgari PV. Emerging drugs for rheumatoid arthritis. Expert Opin Emerg Drugs. 2008;13:175–96.
Acknowledgments
The authors have a cooperative research and development agreement with Pfizer Inc.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Takatori, H., Kanno, Y., Chen, Z. et al. New complexities in helper T cell fate determination and the implications for autoimmune diseases. Mod Rheumatol 18, 533–541 (2008). https://doi.org/10.1007/s10165-008-0099-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10165-008-0099-z