
Abstract
The voltage-gated potassium channel subunit Kv1.1 encoded by the Kcna1 gene is expressed in many brainstem nuclei, and electrophysiological studies of Kcna1-null mutant (−/−) single neurons suggest that channels containing this subunit are critical for precise processing of rapid acoustic perturbations. We tested the hypothesis that brief offsets of a background noise are behaviorally less salient for Kcna1 −/− mice, measured by changes in noise offset inhibition of acoustic startle reflexes (ASR). In experiment 1, noise offset was followed by ASR-eliciting sound bursts either after 1–10 ms quiet intervals or after the return of noise for 10–290 ms following 10-ms quiet gaps. ASR inhibition to offset and gaps was initially higher in +/+ mice but persisted longer in −/− mice. Experiment 2 contrasted brief abrupt offsets with ramped offsets of the same duration up to 10 ms, the ramps intended to simulate progressively slower internal decays of afferent processing. Both groups had greater inhibition for abrupt offsets at asymptote, and this difference was evident at the 1-ms interval in +/+ but not −/− mice. Further, the asymptotic effect of ramped offsets in +/+ mice was equal to that produced by abrupt offsets in null mutants, suggesting more perseveration of internal afferent activity following noise offset in −/− mice. Overall, these data are consistent with prior electrophysiological studies showing that the neural mechanisms for processing acoustic transients are less effective in Kcna1 −/− mice and support previous proposals that Kv1.1 contributes to the perception of animal vocalizations and human speech.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The low-threshold Kv1.1 potassium-ion channel encoded by the Kcna1 gene in mice is highly expressed in the cells of the spiral ganglion and in the auditory brainstem pathways that are critical for encoding, processing, and transmitting rapid acoustic perturbations with minimal temporal distortion (Adamson et al. 2002; Grigg et al. 2000; Rosenberger et al. 2003). The brainstem neural structures include monaural pathways that begin with the octopus cells of the posterior ventral cochlear nucleus (VCN) that continue through the superior olivary complex (SOC) and into the midbrain that may contribute to temporal pattern recognition (Bal and Oertel 2001) and the monaural-to-binaural cascades that begin in the bushy cells of the VCN and continue into other nuclei of the SOC that process the interaural time and level cues necessary for sound localization (Trussell 2002). The unique signature of these neurons is their high fidelity of transmission as embodied in their remarkably small standard deviations in first spike latencies (“jitter”) to acoustic stimuli: at best 50–100 μs in octopus cells (Rhode and Smith 1986) and, on average, 200 μs in bushy cells (Kopp-Scheinpflug et al. 2003). The functional importance of the Kv1.1 subunit of Kv1 ion channels for these neurons is documented in the loss of the one-to-one correspondence of their outputs to inputs and an increased onset jitter when these channels are blocked by neurotoxins and examined with in vitro electrophysiology (Manis and Marx 1991; Brew and Forsythe 1995; Golding et al. 1999; Bal and Oertel 2001), or if cells from Kcna1 −/− null mutant mice are studied in either in vitro or in vivo electrophysiology (Brew et al. 2003; Gittelman and Tempel 2006; Kopp-Scheinpflug et al. 2003).
These earlier experiments have focused on the temporal precision of onset encoding, but the same neurons must also keep track of the offsets of acoustic stimuli, by the rapid cessation of firing at stimulus offset in some nuclei and the timing of offset spikes in others (Kadner and Berrebi 2008; Kopp-Scheinpflug and Forsythe 2011; Felix et al. 2011), and Kv1.1 subunits may contribute to these features as well. Indeed an unusually long delay of offset responding that lasted for several seconds has been seen in Kcna1 −/− cells of the caudal nucleus of the solitary tract with in vitro recording in mice (Kline et al. 2005), but comparable effects not been examined with acoustic stimuli.
Here we examine this hypothesis in Kcna1 null mutant (−/−) and wild-type (+/+) mice in two related behavioral tests using the inhibition of a probe acoustic startle reflex (ASR) to assess the effects of noise offsets and brief gaps in an otherwise continuous background noise. The first experiment was designed with one set of conditions to determine the time course of the induction of inhibition of the probe ASR when its eliciting stimulus (ES) was presented after an abrupt noise offset at brief intervals up to 10 ms and then described the time course of the recovery from inhibition. The second experiment was focused on the induction of inhibition, comparing the behavioral effects of abrupt noise offsets with those of ramped noise offsets. The intent of this manipulation was to provide an external stimulus condition that might simulate the internal decay of afferent activity following an abrupt noise offset. It was patterned on a psychophysical procedure developed by Békésy (1933/1960) and then used by Miller (1948) to measure the rate of decline of sensory activity in humans at stimulus offset. Their idea was that if a stimulus with an abrupt offset could be distinguished from stimuli with relatively gradual slopes that lasted for some critical duration and not distinguished from stimuli having more acute slopes, then the persistence of the “physiological decay” of the internal representation of an abrupt offset must equal that of the critical duration of the external ramp.
Materials and methods
Subjects
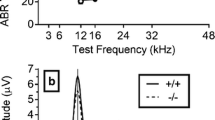
C3HeB.129S7(B6)-Kcna1 tm1Tem of mice were used in this experiment, 29 Kcna1 wild-type (+/+) and 24 Kcna1 null mutant (−/−). An additional 7 +/+ mice and 6 −/− mice were eliminated because of their having a poor ASR (five of the 13 eliminated mice were littermates). All mice were born and raised in the vivarium at the University of Rochester from Kcna1 heterozygote (+/−) breeding stock obtained from The Jackson Laboratory (Bar Harbor, ME, USA). DNA was extracted from tail snips at about post-natal day (PND) 18, and mice were genotyped using the protocol described at http://depts.washington.edu/tempelab/Protocols/KCNA1.html. The ambient noise level in the colony room was 40 dB SPL at 2 kHz and decreased linearly to 25 dB SPL at 24 kHz on a log-frequency scale. Prior to experimentation, mice were screened for obstructions of the external auditory meatus, and for about half of these mice, peripheral hearing sensitivity was assessed via a screening test for hearing (the auditory brainstem response (ABR) test) on about PND 18. In the colony as a whole, a total of 94 −/− mice and 151 +/+ mice have received this ABR test. The overall mean threshold differed between the two populations by less than 1 dB SPL over the standard test frequencies of 3, 6, 12, 16, 24, 36, and 48 kHz (see Table 1). All procedures were approved by the University of Rochester Committee on Animal Resources, in accord with Public Health Service regulations and the Federal Animal Welfare Act.
Apparatus
The experimental apparatus was contained in an anechoic chamber (inside dimensions, 84 × 84 × 84 cm, manufactured by the Eckel Corp., Cambridge MA, USA; model number 555-250-3), and the chamber was housed within a sound attenuating room (IAC, Bronx, NY, USA). The mice were tested one at a time in a test cage, oval in shape and 5 cm wide, 7 cm long, and 4 cm high that was constructed of wire to be transparent to acoustic stimulation. It was mounted on a suspended acrylic platform to which an accelerometer (Model SA-2-300, Statham Laboratories, Beverly Hills, CA, USA) was attached. The output of the accelerometer was amplified then routed to an A/D converter (TDT RP2) and recorded continuously, and the response to the acoustic startle reflex ES was integrated using custom software over a 100-ms interval beginning at ES onset (this providing the ASR measure). In experiment 2, a prestimulus period of 100 ms was also recorded on every trial to assess restless activity prior to the ES (the ACT measure). The ES was a 110-dB (peak-to-peak, SPL linear scale) 20-ms duration noise burst with ~0 ms rise/fall times, provided by a real-time digital processor (TDT RP2) routed through a programmable attenuator (TDT PA5) then amplified and delivered to a Yamaha Professional Series Compression tweeter. The spectrum of the ES varied by no more than +/− 2 dB over the range of octaves centered at 1 to 32 kHz. The background noise was a 70-dB SPL noise provided by a second real-time digital processor (TDT RP2) routed through a second programmable attenuator (TDT PA5) and delivered to a Panasonic high-frequency leaf tweeter that varied by no more than +/− 6 dB over a range of 2 to 100 kHz. The ambient noise level in the chamber was less than 25 dB SPL for all frequencies above 125 Hz. Sound levels were measured with a ¼ in Bruel and Kjaer microphone, model 4135, connecting to a measuring amplifier Bruel and Kjaer, model 2610. Stimulus presentation and response measurements were under computer control. Figure 1 provides an oscilloscope-like schematic of the background noise stimulus condition and the ES of a control trial (Fig. 1A) used in the present experiments and a representative accelerometer response output on a control trial (Fig. 1C), and a schematic of a particular test stimulus used in both experiments, an abrupt noise offset followed 10 ms later by the ES (Fig. 1B) as well as a representative inhibited response in this stimulus condition (Fig. 1D).

An oscilloscope-like schematic of the background noise stimulus condition and the ES of a control trial (A) used in the present experiments and a representative accelerometer response output on a control trial (C); a schematic of a particular test stimulus used in both experiments, an abrupt noise offset followed 10 ms later by the ES (B) as well as a representative inhibited response in this stimulus condition (D).
Procedures
Experiment 1: The time course of ASR inhibition from noise offset and its recovery
A total of 13 Kcna1 +/+ mice and 7 −/− mice completed this experiment, on average at 31.3 days of age for +/+ mice and 28.9 for −/−. These mice also were used in experiment 2, below, and in three other startle experiments on every other day, one using tonal startle stimuli, a second including a visual prepulse, and a third using a gap prepulse. The mice received baseline control trials on which the ES was presented in the presence of the continuous background noise and two types of test trials: In the induction phase, the ES was presented just after noise offset, the offset leading the ES at intervals of 1, 2, 3, 5, or 10 ms (lead time, LT). In the recovery phase, all of the trials began with the return of the noise after a gap of 10 ms, and the ES was presented in the resumed noise background after different delays: 0, 10, 20, 30, 40, 50, 70, 90, 140, and 290 ms: In effect, the conditions of this phase duplicate those of an experiment intended to describe the time course of inhibition following a 10-ms gap in noise. There were 17 stimulus conditions in all: 2 ES-alone control trials, 14 prestimulus conditions in which the ES was preceded by either noise offset alone or by a gap in noise with different recovery times, and 1 “no-stimulus condition” in which spontaneous ACT was measured over the same 100-ms interval as if an ES had been presented. The stimulus conditions were randomly ordered in 11 blocks comprised of 17 trials each with the order of conditions randomized within blocks and an intertrial interval averaging 20 s (range 15–25 s).
Experiment 2: ASR inhibition for abrupt vs. ramped noise offsets
A total of 25 Kcna1 +/+ mice and 23 −/− mice completed this experiment, on average at 32.4 days of age for +/+ mice, 31.2 for −/− mice. The mice received baseline control trials on which the ES was presented in the presence of the continuous background noise and two types of test trials in which the ES was presented just after noise offset, the offset leading the ES at intervals of 1, 2, 3, 4, 5, or 10 ms (LT). On half of these trials the offset was abrupt, with a fall time of 0 ms, and on half of the trials the offset followed a linear down ramp, with the ramp time of the noise being equal to the interval between the onset of the ramp and the ES (1, 2, 3, 4, 5, and 10 ms). Thus, all of the ES presentations in these test trials were presented in quiet: in one set of trials after silent periods varying between 1 and 10 ms all beginning with an abrupt offset and in the other set of trials also in quiet but at the end of the ramp that had a duration between 1 and 10 ms. There were 14 stimulus conditions in total: two ES-alone baseline trials in noise and 12 prestimulus conditions with six lead times for each of the two offset manipulations, abrupt or gradual. There was not an explicit “no-stimulus” condition, the 100-ms measure of spontaneous ACT being taken before each of the ES-alone presentations. These conditions were given in random order within each of 11 blocks having 14 trials, at an average intertrial interval of 20 s.
Data analysis
The mean response measures are given in arbitrary voltage units. The mean of the ASR values for each condition (including the no-stimulus conditions providing the ACT measure) was calculated for each subject, after excluding the first block of trials to avoid possible skewing effects of an initial novelty of the ES. The statistical comparison of the control ASR trials and the “no-stimulus” ACT trials within each mouse was taken as evidence for its expressing a significant startle reaction or not, and each of these two measures alone provided comparisons between +/+ and −/− mice in the strength of their startle reactions and the degree of their restless activity. The excluded mice either did not have a significant difference (p < 0.05) between their control ASR and ACT measures or, if significant, the ASR was not at least twice the value of the ACT. The comparison of the mean ASR on control trials and in each noise offset condition was then used to calculate the strength of inhibition provided by each condition (by convention called prepulse inhibition (PPI); Ison and Hammond 1971). PPI was defined as a ratio of each subject’s mean ASR amplitude in each prestimulus condition (ASRp) compared with its no-prepulse control baseline (ASRc), using the formula PPI = 1 − [ASRp/ASRc].
Repeated-measures analyses of variance (ANOVA) with lead time, recovery time, or offset condition as within-subject variables and genotype as a between-subject variable were performed with SPSS v.18 (IBM-SPSS Inc, Somers, NY, USA). The p values provided by the ANOVA were adjusted via the Hunyh–Feldt method for non-homogeneity of between-cell correlations, and effect sizes were determined by SPSS partial eta-squared measures (\( \eta_{\text{p}}^2 \)). GraphPad Prism software (version 4.2) was used for the graphical presentation of the data and also to calculate non-linear regression functions and supplemental Pearson or Welch’s t tests for specific stimulus conditions (in which case effect sizes are given as R 2 or as Cohen’s d).
Results
Differences in ASR and ACT for Kcna1 +/+ vs. −/− mice were not significant
The average control ASR and ACT amplitudes (summed over experiments for mice that were tested in experiments 1 and 2) were not significantly different, but the ASR amplitudes were higher in the +/+ mice (with the +/+ mice first) for the mean [SEM] of the ASR (1,439 [212] vs. 1,003 [120], Welch’s t (46) = 1.79, p = 0.08, R 2 = 0.07) and the ACT amplitudes were greater in the −/− mice (244 [18] vs. 282 [18], t (54) = 1.46, p = 0.15, R 2 = 0.04). The “near significance” difference for the ASR remains of interest, however, because Karcz (2011) found a significant difference in the mean ASR between binaural Kcna1 +/+ and −/− mice, but no difference when the mice were monauralized with an ear plug. Karcz (2011) had located the ES speaker directly over the head of the mouse, while in the present experiments, the speaker providing the background noise was overhead and the ES speaker was off to the side of the mouse. This would result in the ES being more intense at one ear than the other, which possibly may have been responsible for reducing the ASR difference in the present experiments.
Experiment 1: Less noise offset inhibition and slower ASR recovery in Kcna1 −/− mice
Figure 2 provides the data for the induction of inhibition and then its recovery provided by noise offset in experiment 1. In the induction phase, both +/+ and −/− groups showed a rapid development of inhibition, though the +/+ mice were slightly faster, and also provided higher asymptote levels of inhibition. In the recovery phase, both groups had a relatively slow return to baseline from their peak of inhibition, but the −/− mice had a slower rate of recovery than the +/+ mice. The ANOVA of the induction phase provided a significant effect for genotype: F(1/18) = 4.898, p = 0.04, \( \eta_{\text{p}}^2 \) = 0.21 and for LT: F(4/72) = 21.211, p < 0.001, \( \eta_{\text{p}}^2 \) = 0.54 and a significant interaction between genotype and the cubic trend of LT: F(4/72) = 6.288, p < 0.022, \( \eta_{\text{p}}^2 \) = 0.26. The depicted best-fit one-phase exponential association to these data provided a significant difference in the asymptotic levels of inhibition, the Y max [95% confidence interval], for +/+ = 0.64 [0.56, 0.71] and for −/− = 0.48 [0.34,0.61] and a half-life faster in the +/+ mice, but not significantly so, for the half-life [95% CI], for +/+ = 1.39 [1.05,2.08] and for −/− = 1.68 [0.99, 6.41]. In the recovery phase, the ANOVA provided a significant effect for LT: F(8/144) = 17.863, p = <0.001, \( \eta_{\text{p}}^2 \) = 0.50, a significant interaction between LT and genotype: F(8/144) = 4.261, p < 0.001, \( \eta_{\text{p}}^2 \) = 0.20, but not a significant main effect for genotype, F = 0.43. The depicted best-fit one-stage exponential decay for the +/+ mice provided a plateau (95% CI) of −0.05 (−0.17, 0.06) and a half-life (95% CI) of 55 (39, 95) and for the −/− mice a plateau of 0.14 (−.04, 0.31). However, if given a recovery time as long as the 15–25-s intertrial interval, then the ASR must by definition return to the baseline control level and thus at some interval the plateau in both groups must be the same. It seems more relevant to this comparison of the two groups that Figure 2 shows that PPI in the +/+ mice reached the baseline within the 290-ms recovery period while the −/− mice did not: At 290 ms, their residual PPI was above 0, t(6) = 2.243, one-tail p = 0.033, Cohen’s d = 0.85.

Exp. 1: On the left, the induction phase showing the development of response inhibition (Mean and SEM) provided by the offset of the background noise occurring in lead times of 1 to 10 ms before the startle eliciting stimulus; and on the right, the recovery phase beginning with the onset of the noise at the end of a 10 ms gap, at lead times of 0 to 290 ms before the startle eliciting stimulus. At C is the control baseline with no prior noise-offset. The filled symbols and the best-fit solid one-phase growth and decay functions describe the behavior of the Kcna1 +/+ mice, the open circles and dashed lines describe the behavior of the Kcna1 −/− mice.
More rapid development and higher levels of noise offset inhibition in the +/+ mice
Figure 3A provides the mean (SEM) PPI data in each group, separately for the abrupt noise offset and the ramped noise offset. The strength of inhibition rapidly increased in both stimulus conditions for both groups, but overall there was more inhibition for the abrupt offset compared to the ramped offset and more inhibition in the +/+ compared to the −/− mice. Of additional interest is the observation that at the 1-ms interval, the inhibitory effect of the abrupt offset was significantly greater than that of the ramped offset in the +/+ mice, while at this very brief interval, the two stimulus conditions provided identical levels of inhibition in the −/− mice. This differences between the groups can be seen more clearly in Figure 3B, which depicts the mean (SEM) difference scores between the two stimulus conditions in +/+ and −/− mice: In the +/+ group, the difference between the abrupt and ramped stimulus conditions was fully expressed at the 1-ms interval, while in the −/− mice, this difference more slowly developed between 1 and 4 ms. The ANOVA of the data depicted in Figure 3A provided significant main effects for genotype: F(1/47) = 16.991, p < 0.001, \( \eta_{\text{p}}^2 \) = 0.266; for the offset condition: F(1/47) = 67.74, p < 0.001, \( \eta_{\text{p}}^2 \) = 0.59; and for LT: F(5/235) = 6.013, p < 0.001, \( \eta_{\text{p}}^2 \) = 0.56. Also there were significant interactions between the offset condition and genotype: F(1/47) = 6.56, p < 0.014, \( \eta_{\text{p}}^2 \) = 0.12 and between offset condition, genotype, and the linear trend for LT: F(1/47) = 6.328, p < 0.015, \( \eta_{\text{p}}^2 \) = 0.12.

Exp. 2: Above (A), depicts the development of inhibition (Mean, SEM) for abrupt offsets (circles) and ramped offsets (diamonds) with increasing lead time, 1 to 10 ms, between the noise offset or the beginning of the offset ramp and the ES, and C as the baseline control condition. The solid symbols and solid best-fit growth functions describe the behavior of the Kcna1 +/+ mice, the open symbols and dotted lines the Kcna1 −/− mice. Below (B), the development of the difference between the abrupt and the ramped offsets for the two groups, the asymptotic difference apparent at just 1 ms in the +/+ mice, and a more gradual rise to about the same asymptotic difference only after 4 ms in the −/− mice.
Inhibition for abrupt offsets in −/− mice was matched by ramped offsets in +/+ mice
One notable feature of the data described in Figure 3A is the observation that the behavioral effectiveness of the ramped offsets for the +/+ mice approximated that of the abrupt offset in the −/− mice, the ANOVA for this comparison providing F < 1. The best-fit one-phase exponential regression lines depicted in Figure 3A showed that temporal development of inhibition for the abrupt offset in the +/+ mice was significantly faster than that of all other conditions, which did not differ among themselves: for the half-life of the +/+ abrupt data, mean [95% CI] = 0.88 [0.72, 1.15] compared to the +/+ ramped data, mean [95% CI] = 1.42 [1.08, 2.04]; to the −/− abrupt data, mean [95% CI] = 1.34 [0.99, 2.07]; and to the −/− ramped data, mean [95% CI] = 1.31 [0.92, 2.24].
Discussion
The task of this project was to determine if the Kv1.1 subunit of the Kv1 ion channel that is encoded by the Kcna1 gene contributes to the neural processing of acoustic offsets, as has been demonstrated by the functional deficits seen in the responses of single cells of the Kcna1 null mutant mouse to acoustic onsets in electrophysiological studies in vivo (Kopp-Scheinpflug et al. 2003; Karcz et al. 2011) and to current injections in vitro (Brew et al. 2003; Gittelman and Tempel 2006). The behavioral phenomenon we studied was the very rapid development of a profound reduction in the amplitude of the ASR immediately following the offset of an otherwise continuous noise, a phenomenon previous noted in rats (Stitt et al. 1973; Ison 1982) and in gerbils (Ison et al. 2005) as well as in mice (Ison et al. 1998; Ison and Allen 2003). This behavioral effect of noise offset was reduced in Kcna1 −/− mice in both its asymptotic level and its rate of growth. In addition, the behavioral difference between abrupt and ramped offsets was less evident in −/− mice: For the +/+ mice, the behavioral difference between the two offset conditions was already at its maximum by 1 ms, while the difference only emerged over the next 3 ms in the −/− mice. Further, there was a near equivalence between the growth functions for the ramped offset in +/+ mice and the abrupt offset in −/− mice. This equivalence supports the theoretical conclusion that the persisting post-stimulus afferent activity following the abrupt offset in the −/− mice approximated the level of the externally driven afferent activity during the offset ramps in the +/+ mice.
We note that the specific perceptual hypothesis of Békésy and of Miller also took the form of a comparison between external offset ramps and either the physiological or sensory passive decay of afferent stimulation after abrupt offsets, but the neural basis of the behavioral phenomenon seen here is not certain. Qualitatively, save for its very much faster time course and presentation of much more intense probe stimuli, the loss of reflex strength appears as a sort of post-excitatory depression similar to the classic finding of recovery of auditory nerve firing to probe tones after stimulus offset observed in the cat (Young and Sachs 1973) and in the post-stimulus recovery of detection of probe tones by human listeners in the psychophysical laboratory (Sachs and Young 1973); or perhaps more directly pertinent to the present work, our data are similar to the activity-driven synaptic depression and recovery at the endbulb of Held in the ventral cochlear nucleus of the mouse (Wang and Manis 2008). But curiously in contrast to these two examples, startle responses are not depressed in the presence of moderate levels of background noise but only immediately after their offset. And while caution is appropriate in proposing direct homologies for mouse behavior and human perception, neither the presence of noise nor the presentation of noise offsets at brief intervals seems to have any noticeable effect on the apparent intensity of the startle ES for the human listener and perhaps also no effect on its sensory impact for the mouse.
For example, given the identical stimuli provided to the mouse in the free field in a two-alternative forced-choice task with pairs of ES presentations, one with a preceding abrupt noise offset and one without, a single observer (JRI) could accurately identify the stimuli and also hear the silent period prior to the ES for offsets with lead times of 80 ms and more. But as lead times dropped to 50 ms, then performance fell to chance levels, this revealing the not surprising masking of the 70-dB offset by the immediately following 110-dB noise pulse but also the lack of any obvious difference in the apparent intensity of the ES presented in these two conditions. Also, in other trials with no presentation of the ES, there was 100% detection of 10-ms gaps. We thus infer that while the effects of the abrupt offsets and the particular durations of ramped offsets used in the second experiment with mice must certainly depend on their differentially affecting particular afferent pathways, these pathways probably are not directly available to introspection because of the masking effect of the noise ES. In the absence of the noise pulse, these same ramped manipulations become variables in a standard gap detection protocol, and in gap detection experiments with human listeners, these brief ramps are not heard as such, but instead they affect the probability of the listener’s detecting the gap (Allen et al. 2002).
These observations suggest that rather than passive decay, there is an active neural mechanism encoding noise offset that develops its inhibitory effect on a sub-millisecond scale in Kcna1 +/+ mice and is as sensitive to offset decay times as other cells are sensitive to onset rise times. Further, this circuit must be in close proximity to the startle reflex pathway, described, e.g., by Koch (1999), in order to effectively inhibit the expression of a response that has an onset latency as measured in the EMG of about 6 ms in the forelimb muscles of the rat (Hammond et al. 1972). Kadner and Berrebi (2008), Felix et al. (2011), and Kopp-Scheinpflug and Forsythe (2011) have observed that the cessation of stimulus-evoked neural activity in the medial nucleus of the trapezoid body drives rebound offset responses in the superior pariolivary nucleus. We suggest that these nuclei that are part of a complex system known to be sensitive to stimulus rise time (McGinley and Oertel 2006) and also are known to express Kcna1 may be critical parts of the afferent chain responsible for the behavioral effects of noise offsets observed in the present experiments.
They are likely also to participate in the perception of brief gaps in noise in the more common “standard” gap experiment described above, as suggested by Kadner and Berrebi (2008), Felix et al. (2011), and Kopp-Scheinpflug and Forsythe (2011) and as shown by Allen et al. (2008) who reported a deficit in asymptotic inhibition in Kcna1 −/− mice for suprathreshold gap durations. But for gap detection, these relatively caudal nuclei must certainly act in concert with more rostral mechanisms, as is shown by clinical observations that bilateral lesions of auditory cortex substantially inflate auditory gap detection thresholds in human patients (Buchtel and Stewart 1989). The same effect on gap detection is found in rats following bilateral ablation of the auditory cortex (Bowen et al. 2003) and spreading depression-induced functional decortication (Ison et al. 1991), but it is significant that these manipulations do not at all affect ASR inhibition provided by noise offset
References
Adamson CL, Reid MA, Mo Z-L, Bowne-English J, Davis RL (2002) Firing features and potassium channel content of murine spiral ganglion neurons vary with cochlear location. J Comp Neurol 447:331–350
Allen PD, Virag TM, Ison JR (2002) Humans detect gaps in broadband noise according to effective quiet time without additional cues from abrupt envelope changes. J Acoust Soc Am 112:2967–2974
Allen PD, Schmuck N, Ison JR, Walton JP (2008) Kv1.1 channel sub-units are not necessary for high temporal acuity in behavioral and electrophysiological gap detection. Hear Res 246:52–58
Bal R, Oertel D (2001) Potassium currents in octopus cells of the mammalian cochlear nucleus. J Neurophys 86:2299–2311
Békésy G (1933/1960) Experiments in hearing. McGraw-Hill, New York
Bowen GP, Taylor MK, Lin D, Ison JR (2003) Auditory cortex lesions impair both temporal acuity and intensity discrimination in the rat, suggesting a common mechanism for sensory processing. Cereb Cortex 13:815–822
Brew HM, Forsythe ID (1995) Two voltage-dependent K+ conductances with complementary functions in postsynaptic integration at a central auditory synapse. J Neurosci 15:8011–8022
Brew HM, Hallows J, Tempel BL (2003) Hyperexcitability and reduced low threshold potassium currents in auditory neurons of mice lacking the channel subunit Kv1.1. J Physiol 548:1–20
Buchtel HA, Stewart JD (1989) Auditory agnosia: apperceptive or associative disorder? Brain Lang 37:12–25
Felix RA II, Fridberger A, Leijon S, Berrebi AS, Magnusson AK (2011) Sound rhythms are encoded by postinhibitory rebound spiking in the superior paraolivary nucleus. J Neurosci 31:12566–12578
Gittelman JX, Tempel BL (2006) Kv1.1-containing channels are critical for temporal precision during spike initiation. J Neurophysiol 96:1203–1214
Golding NL, Ferragamo MJ, Oertel D (1999) Role of intrinsic conductances underlying responses to transients in octopus cells of the cochlear nucleus. J Neurosci 19:2897–2905
Grigg JJ, Brew HM, Tempel BL (2000) Differential expression of voltage-gated potassium channel genes in auditory nuclei of the mouse brainstem. Hear Res 140:77–80
Hammond GR, McAdam DW, Ison JR (1972) Effects of prestimulation on the electromyographic response associated with the acoustic startle reaction in rats. Physiol Beh 8:535–537
Ison JR (1982) Temporal acuity in auditory function in the rat: reflex inhibition by brief gaps in noise. J Comp Physiol Psych 96:945–954
Ison JR, Allen PD (2003) A diminished rate of “physiological decay” at noise offset contributes to age-related changes in temporal acuity in the CBA mouse model of presbycusis. J Acoust Soc Am 114:522–528
Ison JR, Hammond GR (1971) Modification of the startle reflex in the rat by changes in the auditory and visual environments. J Comp Physiol Psych 75:435–452
Ison JR, O’Connor K, Bowen GP, Bocirnea A (1991) Temporal resolution of gaps in noise by the rat is lost with functional decortication. Behav Neurosci 105:33–40
Ison JR, Agrawal P, Pak J, Vaughn WJ (1998) Changes in temporal acuity with age and with hearing impairment in the mouse: a study of the acoustic startle reflex and its inhibition by brief decrements in noise level. J Acoust Soc Am 104:1696–1704
Ison JR, Moore JT, Rivoli PJ, and Allen P (2005) Behavioral measures of complex auditory processing in the adult Mongolian gerbil (Meriones unguiculatus). Soc Neurosci Abstr 31: #44.20
Kadner A, Berrebi AS (2008) Encoding of temporal features of auditory stimuli in the medial nucleus of the trapezoid body and superior paraolivary nucleus of the rat. Neurosci 151:868–887
Karcz A (2011) The significance of the low voltage-gated potassium channel subunit Kv1.1 for the processing of sound source location. Ph.D. dissertation, University of Leipzig
Karcz A, Henning MH, Robbins CA, Tempel BL, Rübsamen R, Kopp-Scheinpflug C (2011) Low-voltage activated Kv1.1 subunits are crucial for the processing of sound source location in the lateral superior olive in mice. J Physiol 589:1143–1157
Kline DD, Bunuel MC, Glazebrook P, Pang I, Ramirez-Navarro A, Prebake NR, Kane DL (2005) Kv1.1 deletion augments the afferent hypoxic chemosensory pathway and respiration. J Neurosci 25:3389–3399
Koch M (1999) The neurobiology of startle. Prog Neurobiol 59:107–128
Kopp-Scheinpflug C, Forsythe ID (2011) The sound of silence: ionic mechanisms encoding sound termination. Neuron 71:911–925
Kopp-Scheinpflug C, Fuchs K, Lippi WR, Tempel BL, Rübsamen R (2003) Decreased temporal precision of auditory signaling in Kcna1-null mice: an electrophysiological study in vivo. J Neurosci 23:9199–9207
Manis BP, Marx SO (1991) Outward currents in isolated ventral cochlear nucleus neurons. J Neurosci 11:2865–2880
McGinley MJ, Oertel D (2006) Rate thresholds determine the precision of temporal integration in principal cells of the ventral cochlear nucleus. Hear Res 216–217:52–63
Miller GA (1948) The perception of short bursts of noise. J Acoust Soc Am 20:160–170
Rhode WS, Smith PH (1986) Encoding timing and intensity in the ventral cochlear nucleus of the cat. J Neurophysiol 56:261–286
Rosenberger MH, Fermium T, Cassidy JH, Covey E (2003) Expression of the Kv1.1 ion channel subunit in the auditory brainstem of the big brown bat, Eptesicus fuscus. J Comp Neurol 462:101–120
Sachs MB, Young ED (1973) Recovery of detection probability following sound exposure: comparison of physiology and psychophysics. J Acoust Soc Am 54:1544–1553
Stitt CL, Hoffman HS, Marsh R (1973) Modification of the rat’s startle reaction by termination of antecedent acoustic signals. J Comp Physiol Psychol 84:207–215
Trussell LO (2002) Cellular mechanisms for information coding in auditory brainstem nuclei. In: Oertel D, Fay RR, Popper AN (eds) Integrative functions in the mammalian auditory pathway. Springer, New York, pp 72–98
Wang Y, Manis BP (2008) Short-term synaptic depression and recovery at the mature mammalian endbulb of Held synapse in mice. J Neurophysiol 100:1255–1264
Young E, Sachs MB (1973) Recovery from sound exposure in auditory-nerve fibers. J Acoust Soc Am 54:1535–1543
Acknowledgments
This research was supported by US Public Health Service NIH grants AG0095247 and DC05409 and by the Schmitt Program in Integrative Brain Research. We thank Bruce Tempel, Helen Brew, and two anonymous reviewers for helpful comments on an earlier version of this manuscript; Dr. Robert Frisina, Xiaoxia Zhu, and Martha Zettel for genotyping the mice; and Dr. William O’Neill, Dr. Joseph Walton, and John Housel for their contributions to ABR testing.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ison, J.R., Allen, P.D. Deficits in Responding to Brief Noise Offsets in Kcna1 −/− Mice Reveal a Contribution of This Gene to Precise Temporal Processing Seen Previously Only for Stimulus Onsets. JARO 13, 351–358 (2012). https://doi.org/10.1007/s10162-011-0312-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10162-011-0312-1