
Abstract
Background
Recently, comprehensive genetic approaches for steroid-resistant nephrotic syndrome (SRNS) using next-generation sequencing (NGS) have been established, but causative gene mutations could not be detected in almost 70% of SRNS patients. Main reason for the low variant detection rate is that most of them are SRNS caused not by genetic but by immunological factors. But some of them are probably because of the difficulty of detecting copy number variations (CNVs) in causative genes by NGS.
Methods
In this study, we performed two analytical methods of NGS data-dependent pair analysis and custom array comparative genomic hybridization (aCGH) in addition to NGS analysis in an infantile nephrotic syndrome case.
Results
We detected only one known pathogenic heterozygous missense mutation in exon 7 of COQ6 c.782C > T, p.(Pro261Leu) by NGS. With pair analysis, heterozygous exon 1–2 deletion was suspected and was confirmed by custom aCGH. As a result, a small CNV was successfully detected in the COQ6 gene. Because we could detect variants in COQ6 and could start treatment by coenzyme Q10 (CoQ10) in his very early stage of SRNS, the patient achieved complete remission.
Conclusions
These relatively novel methods should be adopted in cases with negative results in gene tests by NGS analysis. Especially, in cases with CoQ10 deficiency, it is possible to delay initiating dialysis by starting treatment at their early stages.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Comprehensive genetic analysis using next-generation sequencing (NGS) of causative genes for steroid-resistant nephrotic syndrome (SRNS) has been widely established globally [1,2,3]. It enables us to treat certain monogenic SRNS patients with specific therapies, such as coenzyme Q10 (CoQ10) supplementation for patients with CoQ10 deficiency-related gene variants, including PDSS2, COQ2, COQ6, and ADCK4, exhibiting so-called CoQ10 glomerulopathies [4].
CoQ10 is a ubiquitous lipophilic substance that is biosynthesized in mitochondria and plays essential roles in adenosine triphosphate production through the mitochondrial respiratory chain, beta-oxidation of fatty acids, and pyrimidine biosynthesis [5]. Another important role of CoQ10 is modulating cellular oxidant stress. At least 15 genes have been reported to be causative of CoQ10-related diseases [6]. Among these, some genes have also recently been reported to be causative of SRNS [7]. These include COQ2 variants, which were detected for the first time in CoQ10 glomerulopathies [8]. The main extra-renal symptoms were nervous system involvement such as encephalopathy and myopathy. Nephropathy was also reported, which usually occurs at an age of under 1 year old and progresses to end-stage renal disease [9]. ADCK4 variants were also reported as being causative of CoQ10 glomerulopathies, but extra-renal symptoms are seldom reported in such cases [10]. The onset of nephrotic syndrome is usually in adolescence [11]. Moreover, in the Asian population with SRNS, it was reported that the rate of patients having ADCK4 gene variants seemed to be higher than that found elsewhere [2].
Comprehensive NGS analysis has revealed that about 30% of SRNS patients possess a monogenic gene variant in genes causative of SRNS; however, comprehensive NGS analysis has the disadvantage of having difficulty detecting copy number variations (CNVs). CNVs are present throughout the human genome and are considered to be a significant source of human genetic variation. They have also been reported to cause diseases [12].
In this study, we conducted NGS analysis on an infantile SRNS patient; moreover, to detect any CNV in this subject, we performed NGS-data-based pair analysis and custom array comparative genomic hybridization (aCGH). Based on the results obtained by these analyses, we also initiated CoQ10 treatment in this case and assessed its efficacy.
Materials and methods
Genetic analysis
A list of all of the genes screened for being causative of SRNS in this study is presented in Supplementary Table S1. Genomic DNA was extracted from peripheral blood leukocytes of the patient and his parents with the QuickGene Mini 80 system (Wako Pure Chemical Industries, Ltd., Tokyo, Japan), in accordance with the kit’s protocol. The patient sample was prepared for targeted sequencing with a HaloPlex target enrichment system kit (Agilent Technologies, Santa Clara, CA), in accordance with the kit’s protocol. Approximately 225 ng of genomic DNA was extracted, subjected to a restricted reaction for fragmentation, and hybridized with NGS probes at 54 °C for 16 h.
Subsequently, the patient’s genomic DNA was amplified by polymerase chain reaction and sequenced using the MiSeq platform (Illumina, San Diego, CA). After completing the NGS analysis, we analyzed the data with SureCall 4.0, by which we can combine the data with the algorithms of alignment NGS data analysis (Agilent Technologies).
Pair analysis
We conducted NGS-data-based CNV analysis, in other words, pair analysis, using SureCall. This pair analysis involved the following five steps. First, the read depth was calculated for each base-pair of targeted regions using the NGS analysis data. Second, the targeted region was divided into 125-base-pair fragments. Third, the average read depth of each fragment was calculated. Fourth, the read depths were normalized with the total read depths and the sample specimen was compared with the reference one. As references, we selected control samples that had been analyzed for gene mutations using the same protocol of NGS, in which disease-causing variants other than those in the targeted regions were detected, and for which there was a small possibility of CNVs occurring at the targeted regions. Fifth, log2[sample read depth (normalized)/reference read depth (normalized)] was calculated. Deletion sites have scores of about − 1.0 for this, while duplication sites have scores of about 1.58, in autosomal genes.
Custom aCGH
Before we conducted custom aCGH, we selected genes for analysis. Here, we selected the COQ6 gene and constructed probes for it and the regions surrounding it. We used a custom HD-CGH Microarray, 8 × 15K (Agilent Technologies), in accordance with the kit’s protocol. Using 1 µg of genomic DNA from a reference human control (Promega) and from the patient, we obtained fragments by heating at 95 °C for 10 min.
Next, we used the SureTag Complete Labeling Kit (Agilent Technologies) to label fragmented DNA of the reference sample with Cy3 and of the patient sample with Cy5 fluorescent dUTP. Next, we conducted purification of the samples and removed the unincorporated nucleotides and dyes using purification columns (Agilent). After purification, we obtained the labeled fragmented samples along with human Cot-1 DNA and hybridized them on array slides. Labeled samples were hybridized to custom CGH arrays with heating at 65 °C and 20 rpm for 24 h in a hybridization oven. The slides were scanned by Agilent Microarray Scanner System (Agilent Technologies) at 3-µm resolution. We used Agilent CytoGenomics software (Agilent Technologies) to analyze chromosomal patterns within the microarray profiles.
Semi-quantitative polymerase chain reaction (PCR)
We conducted semi-quantitative PCR to detect a large deletion of exon 1 in COQ6 on the paternal allele using capillary electrophoresis (2100 Bioanalyzer; Agilent Technologies). The concentration of genomic DNA was approximately 50 ng/µL, and PCR was performed with primers covering the regions of exon 1 and exons 6–7 in COQ6 and exon 12 in NUP107 as an internal control. The quantity of PCR products was calculated using the Agilent DNA1000 kit (Agilent Technologies).
Quantitative polymerase chain reaction (qPCR)
We conducted quantitative PCR to detect a large deletion of exon 1 in COQ6 on the paternal allele. SYBR® green real-time RT-PCR amplification was performed using a 7500 Fast Real-time PCR system (Applied Biosystems, Foster City, CA). Real-time PCR assays were carried out in a final volume of 20 µL, consisting of 10 µL of Thunderbird® SYBR® qPCR Mix (Toyobo, Osaka, Japan), 0.04 µL of 50 × ROX reference dye, 6 pmol specific primers for the COQ6 gene, and 10 ng of the sample DNA. The real-time PCR conditions were as follows: 1 cycle for 20 s at 96 °C, followed by 40 cycles of 2 s at 96 °C for denaturation, 15 s at 60 °C for annealing, and 15 s at 72 °C for extension. Spectral data were captured and analyzed using 7500 Real-Time Analysis Software version 2.0.3 (Applied Biosystems, Foster City, CA). All samples were run in triplicate. GAPDH DNA was analyzed as an endogenous copy number reference gene.
Results
Patient
The patient was a 1-year-old male. After birth, he was diagnosed with tetralogy of Fallot. When he was admitted for intracardiac repair at 10 months old, severe proteinuria and hypoalbuminemia (urine protein creatinine ratio of 50 g/gCr and serum albumin of 1.2 g/dL) were detected. He was diagnosed with infantile nephrotic syndrome, for which treatment with steroid was initiated. However, this therapy was not effective and he was diagnosed with SRNS, after which the steroid was tapered and angiotensin-converting enzyme inhibitor (ACE-I) and angiotensin 2 receptor blocker (ARB) were started. At the same time, we decided to conduct a genetic test on the patient. He also underwent auditory brain-stem response (ABR) screening and was shown to have no hearing loss at 1 year old.
Pathological findings
The patient underwent a kidney biopsy; images of the obtained pathological findings are shown in Fig. 1. On light microscopy, the glomeruli showed no remarkable changes, while the results of immunofluorescence staining were all negative. However, electron microscopic examination revealed abnormal proliferation of dysmorphic mitochondria in podocytes, diffuse foot process effacement, and microvillous transformation of podocytes (Fig. 1).

Electron microscopy images of the patient. a In the podocytes, the abnormal proliferation of swollen mitochondria was observed. b Foot process of podocytes showed diffuse effacement. Microvillous transformation was prominent
Genetic analysis
Targeted sequencing with NGS showed one known pathogenic heterozygous missense mutation in exon 7 of COQ6 [c.782C > T, p.(Pro261Leu)] [13]. We also conducted Sanger sequencing and detected the same mutation in both the patient and his mother (Supplemental Fig. 1).
Pair analysis
Next, we conducted pair analysis using the SureCall application. The log ratio of exon 1 in COQ6 was − 1.465 and that of exon 2 in COQ6 was − 1.679 (Supplemental Fig. 2). We thus suspected that the patient had a large heterozygous deletion including exons 1 and 2 of COQ6.
Custom aCGH
By employing custom aCGH, a large heterozygous deletion including the first half of the COQ6 gene was successfully detected (Fig. 2).

Custom aCGH results of the patient. We conducted custom aCGH in the patient using primers for a region including the COQ6 gene. We detected a large deletion, including part of the COQ6 gene. The deleted area extended from exon 1 to the middle of the COQ6 gene
Semi-quantitative PCR
The results of semi-quantitative PCR showed that, in their genomic DNA, the patient and his father had about half the level of exon 1 of COQ6 compared with the levels in the patient’s mother and the control. However, the levels of exons 6 and 7 in COQ6 were the same (Supplemental Fig. 3).
qPCR
The results of qPCR showed that the patient and his father had about half the levels of exon 1 of COQ6 compared with the levels in the patient’s mother and the control (Supplemental Fig. 4). These results indicated that the patient had a compound heterozygous mutation involving deletion of the paternal allele and a known pathogenic missense mutation in the maternal allele.
Effect of CoQ10 supplementation
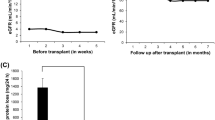
After detecting the COQ6 gene defect, the patient was started on CoQ10 supplementation in addition to ACE-I/ARB. As a result, his proteinuria gradually improved and eventually disappeared. His serum albumin level also increased (Fig. 3).

Clinical course of the patient after treatment with coenzyme Q10 (CoQ10). After starting CoQ10 supplementation, the proteinuria remarkably improved and finally disappeared. The patient’s serum albumin level gradually increased to the normal range. UPCR urine protein creatinine ratio, ACE-I angiotensin-converting enzyme inhibitor, ARB angiotensin receptor blocker
Discussion
Recently, more than 40 genes causative of SRNS have been reported. NGS analysis has also recently become a standard approach for gene screening. However, NGS cannot always detect CNVs, so another approach is needed for their detection. In this study, we conducted NGS, pair analysis, and custom aCGH, enabling the detection of two disease-causing variants, including a large heterozygous deletion in the COQ6 gene, in an infantile SRNS patient.
COQ6 mutations have been reported to be causative of diseases in some patients with inherited nephrotic syndrome; these patients usually exhibit hearing loss [7]. In these patients, proteinuria occurs before the age of 6 years and rapidly develops to end-stage renal disease before 10 years old [14].
CoQ10 glomerulopathies show massive proteinuria, for which immunosuppressant drugs including steroids are not usually effective; most of these patients progress to end-stage renal disease. Recently, CoQ10 has been reported to be an effective therapy for patients with CoQ10 glomerulopathies [10, 14]. Thus, there is currently a growing need for a comprehensive system for screening genes associated with CoQ10 synthesis.
The use of the comprehensive screening system including pair analysis for SRNS patients enabled us to detect CoQ10 glomerulopathy-related genes at early disease stages [14]. In a previous report, it was suggested that early initiation of CoQ10 supplementation is important to prevent progression to end-stage renal disease and worsening neurological involvement in diseases involving CoQ10 deficiency [15]. In line with this, in our patient, CoQ10 was extremely effective because it was started at quite an early stage in the clinical course, at which point there was no hearing loss or glomerulosclerosis.
In a previous report, it was described that all cases with COQ6 defects showed hearing loss and some showed epilepsy or mental retardation [13]. These extra-renal symptoms provide a clue for the diagnosis of CoQ10 glomerulonephropathies. However, it should be noted that cases with ADCK4 variants do not show extra-renal symptoms.
In pathological examinations, the proliferation of abnormally shaped mitochondria in the cytoplasm of podocytes was consistent with the findings in a previous report on CoQ10 glomerulopathies related to COQ6 mutation [13], providing a pathological clue for diagnosing this disease.
To detect CNVs, karyotype analysis for chromosome size variations or in situ hybridization for specific gene variations has conventionally been performed. Recently, array comparative genomic hybridization (aCGH) for detecting genome-wide CNVs or multiplex ligation and probe amplification (MLPA) for detecting specific genes has been conducted [16]; however, commercial aCGH kits can detect CNVs expanded over more than 2–5 kbp, but cannot detect small CNVs. As for commercial MLPA kits, they are only available for a limited number of genes.
NGS-data-based CNV analysis has recently been reported to be an effective method for detecting CNVs; its sensitivity was 92% and specificity was 100% to detect deletions as small as 180 bp and duplications of more than 300 bp in specific targeted gene panels [17]. We also recently published data about the effectiveness of NGS-data-based CNV analysis for inherited kidney diseases [18]. Moreover, custom aCGH has been reported to be effective for detecting CNVs [19]. The main differences between custom aCGH and original whole-genome aCGH are as follows. First, with custom aCGH, we can analyze targeted regions including the candidate genes. Second, we can set probes more densely, every 100 bp, in targeted regions, which would be expected to result in higher sensitivity and specificity to detect CNVs by custom aCGH than by original aCGH.
In the current study, to detect CNVs, we conducted pair analysis using NGS panel sequencing data with SureCall and custom aCGH, which were confirmed by quantitative PCR. All results were clearly concordant, and we concluded that the combination of pair analysis and custom array CGH is an effective approach. However, we know that these approaches are not always available in all clinical settings. In such cases, we recommend the initiation of CoQ10 supplementation even when only heterozygous mutation has been detected by sequencing because we already know the benefits of starting CoQ10 treatment at an early stage. Nonetheless, when the above methods can be applied in combination, the effective detection of CNVs should be possible. The identification of CoQ10 glomerulopathy at an early stage can improve the prognosis of these serious disorders.
Conclusions
The use of a comprehensive gene screening system with NGS was effective to detect causative gene variants in SRNS. In addition, the combination of pair analysis and custom aCGH was remarkably useful to detect CNVs. Specific therapy of CoQ10 for patients carrying COQ6 mutations was very effective when started at an early stage, so it is important not to miss the opportunity to initiate supplementation at an early stage in cases with CoQ10 glomerulopathies.
References
Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26(6):1279–89. https://doi.org/10.1681/ASN.2014050489.
Wang F, Zhang Y, Mao J, Yu Z, Yi Z, Yu L, et al. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2017. https://doi.org/10.1007/s00467-017-3590-y.
Ogino D, Hashimoto T, Hattori M, Sugawara N, Akioka Y, Tamiya G, et al. Analysis of the genes responsible for steroid-resistant nephrotic syndrome and/or focal segmental glomerulosclerosis in Japanese patients by whole-exome sequencing analysis. J Hum Genet. 2016;61(2):137–41. https://doi.org/10.1038/jhg.2015.122.
Doimo M, Desbats MA, Cerqua C, Cassina M, Trevisson E, Salviati L. Genetics of coenzyme q10 deficiency. Mol Syndromol. 2014;5(3–4):156–62. https://doi.org/10.1159/000362826.
Emma F, Bertini E, Salviati L, Montini G. Renal involvement in mitochondrial cytopathies. Pediatr Nephrol. 2012;27(4):539–50. https://doi.org/10.1007/s00467-011-1926-6.
Desbats MA, Lunardi G, Doimo M, Trevisson E, Salviati L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J Inherit Metab Dis. 2015;38(1):145–56. https://doi.org/10.1007/s10545-014-9749-9.
Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol. 2016;12:267. https://doi.org/10.1038/nrneph.2015.214.
Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, Dimauro S, et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 2006;78(2):345–9. https://doi.org/10.1086/500092.
Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18(10):2773–80. https://doi.org/10.1681/ASN.2006080833.
Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Investig. 2013;123(12):5179–89. https://doi.org/10.1172/JCI69000.
Korkmaz E, Lipska-Zietkiewicz BS, Boyer O, Gribouval O, Fourrage C, Tabatabaei M, et al. ADCK4-associated glomerulopathy causes adolescence-onset FSGS. J Am Soc Nephrol. 2016;27(1):63–8. https://doi.org/10.1681/ASN.2014121240.
Girirajan S, Campbell CD, Eichler EE. Human copy number variation and complex genetic disease. Ann Rev Genet. 2011;45:203–26. https://doi.org/10.1146/annurev-genet-102209-163544.
Park E, Ahn YH, Kang HG, Yoo KH, Won NH, Lee KB, et al. COQ6 mutations in children with steroid-resistant focal segmental glomerulosclerosis and sensorineural hearing loss. Am J Kidney Dis. 2017;70(1):139–44. https://doi.org/10.1053/j.ajkd.2016.10.040.
Heeringa SF, Chernin G, Chaki M, Zhou W, Sloan AJ, Ji Z, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Investig. 2011;121(5):2013–24. https://doi.org/10.1172/JCI45693.
Montini G, Malaventura C, Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med. 2008;358(26):2849–50. https://doi.org/10.1056/NEJMc0800582.
Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genom. 2011;12:184. https://doi.org/10.1186/1471-2164-12-184.
Onsongo G, Baughn LB, Bower M, Henzler C, Schomaker M, Silverstein KA, et al. CNV-RF is a random forest-based copy number variation detection method using next-generation sequencing. J Mol Diagn. 2016;18(6):872–81. https://doi.org/10.1016/j.jmoldx.2016.07.001.
Nagano C, Nozu K, Morisada N, Yazawa M, Ichikawa D, Numasawa K, et al. Detection of copy number variations by pair analysis using next-generation sequencing data in inherited kidney diseases. Clin Exp Nephrol. 2018. https://doi.org/10.1007/s10157-018-1534-x.
Vasson A, Leroux C, Orhant L, Boimard M, Toussaint A, Leroy C, et al. Custom oligonucleotide array-based CGH: a reliable diagnostic tool for detection of exonic copy-number changes in multiple targeted genes. Eur J Hum Genet. 2013;21(9):977–87. https://doi.org/10.1038/ejhg.2012.279.
Acknowledgements
This study was supported by a Grant from the Ministry of Health, Labour and Welfare of Japan for Research on Rare Intractable Diseases in the Kidney and Urinary Tract [H24-nanchitou (nan)-ippan-041 to Kazumoto Iijima] in the “Research on Measures for Intractable Diseases” Project; Grants-in-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (subject ID: 17K16262 to Keita Nakanishi, 15K09691 to Kandai Nozu, and 17H04189 to Kazumoto Iijima); and by Japan Agency for Medical Research and Development AMED under Grant number 7930006 to Kandai Nozu and Kazumoto Iijima. We thank Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have nothing to disclose.
Ethical approval
All procedures were reviewed and approved by the Institutional Review Board of Kobe University School of Medicine.
Informed consent
Informed consent was obtained from the patient’s parents.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Nakanishi, K., Okamoto, T., Nozu, K. et al. Pair analysis and custom array CGH can detect a small copy number variation in COQ6 gene. Clin Exp Nephrol 23, 669–675 (2019). https://doi.org/10.1007/s10157-018-1682-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-018-1682-z