
Abstract
The differential diagnosis of acute poststreptococcal glomerulonephritis (APSGN) and idiopathic membranoproliferative glomerulonephritis (MPGN) is sometimes difficult, as they share several key features in their laboratory and histological findings, especially during the acute phase of the diseases. We herein report an idiopathic case of MPGN in which the glomerular deposition of nephritis-associated plasmin receptor (NAPlr), a recently identified nephritic antigen for APSGN, was demonstrated. A 24-year-old postpartum woman developed nephrotic syndrome and hypocomplementemia. Although she showed no apparent findings of a prior infection, her serum titer of antistreptolysin O antibody was elevated. Renal biopsies were performed twice at intervals of 6 months, both of which showed findings fully consistent with those of MPGN. Of note, fluorescent immunostaining for NAPlr was positive in the glomeruli of the first biopsy but not in the second. Despite the use of a corticosteroid, hypocomplementemia persisted for more than 1 year. It was therefore suggested that a streptococcal infection may have influenced the development of glomerular injury in this idiopathic case of MPGN.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Membranoproliferative glomerulonephritis (MPGN) is a type of primary glomerular disease typically showing a chronic clinical course. MPGN is histologically characterized by diffuse mesangial cell proliferation and thickening of the capillary walls due to subendothelial extension of the mesangium [1]. Clinically, patients with MPGN show hypocomplementemia, and some cases have nephrotic syndrome and/or acute nephritic syndrome. MPGN is divided into the idiopathic type of unknown causes and the secondary type that is related to systemic and/or infectious disorders (e.g. hepatitis virus infection, cryoglobulinemia, or systemic lupus erythematosus). The differential diagnosis between acute poststreptococcal glomerulonephritis (APSGN) and idiopathic MPGN is sometimes difficult, as they share several key features in their laboratory and histological findings, especially during the acute phase of the diseases. As most APSGN cases recover without treatment within several months after onset, some cases first diagnosed as APSGN, in which hypocomplementemia persists for more than several months, are usually then diagnosed as idiopathic MPGN.
Recently, the nephritis-associated plasmin receptor (NAPlr), a nephritogenic antigen for APSGN, was isolated and is thought to be useful in the diagnosis of glomerulonephritis associated with streptococcal infection [2–9]. NAPlr is homologous to the group A streptococcus plasmin receptor known as streptococcal glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which is thought to impair glomeruli either directly or indirectly [3, 4]. To date, there have been only a few reports in which NAPlr staining was demonstrated in idiopathic MPGN cases [2, 5]. In this report, we present a case that showed transient immunostaining for the glomerular NAPlr antigen only during the initial phase of the disease, which was finally diagnosed as idiopathic MPGN.
Case report
The patient was a 24-year-old Japanese woman who had received annual medical checkups and had not previously been diagnosed with hypertension or renal insufficiency. When she was pregnant at the age of 23 years, she developed proteinuria at 12 weeks of gestation and hypertension (146/80 mmHg) by the 16th week of gestation, leading to the diagnosis of preeclampsia. Her blood pressure was controlled with the use of hydralazine (60 mg daily), and she delivered a female infant with normal birth weight at 40 weeks and 2 days of gestation. As the hypertension persisted even after the delivery, the dose of hydralazine was increased (90 mg daily) and an angiotensin II receptor blocker (ARB)–diuretic combination drug (candesartan 4 mg and hydrochlorothiazide 6.25 mg daily) was added. Despite these treatments, the proteinuria did not decrease during the postpartum period. At 5 months after the delivery, the patient showed systemic edema and her body weight increased by 5 kg within a week. In the laboratory findings, she had hypocomplementemia (C3 13 mg/dl, C4 13 mg/dl, CH50 18 U/ml), hypoalbuminemia (ALB 1.6 g/dl), and massive proteinuria (14 g/day). She was therefore diagnosed with nephrotic syndrome. Her serum creatinine was slightly elevated (1.5 mg/dl). There was neither physical nor serological evidence of any infections, including hepatitis viruses, human papillomavirus, or any other microorganisms, except for an elevation of the serum titer of antistreptolysin O antibody (ASO) at 218 IU/ml. Serum tests for antinuclear antibodies, antineutrophilic cytoplasmic antibodies, double-stranded DNA antibodies, and cryoglobulins were all negative.
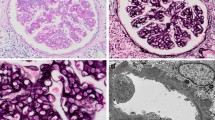
A renal biopsy was performed after obtaining the patient’s informed consent. The renal tissue specimen contained a total of 32 glomeruli, and five of them were globally sclerotic. There were eight glomeruli with cellular crescents. The remaining glomeruli showed mesangial hypercellularity and neutrophil infiltration in the capillary lumens, together with thickening and double-contouring of the basement membrane (Fig. 1a). Fluorescent immunostaining showed granular staining for C3 on the mesangial area and the peripheral capillary walls (Fig. 1c), and staining for immunoglobulin G (IgG), IgA, IgM, and C1q was negative. Of note, immunofluorescent staining for NAPlr using a fluorescein-conjugated mouse monoclonal-specific antibody (Yamasa Corporation, Tokyo, Japan) was positive in the mesangial area (Fig. 1d). According to the ultrastructural evaluations, electron-dense deposits were observed mainly in the mesangial area. Deposits were also found to a lesser extent in the subendothelial spaces, subepithelial spaces, and basement membrane. However, neither humps nor organized deposits were found in the specimen.

Kidney biopsy findings. a Light microscopy findings of the first biopsy [periodic acid methenamine silver (PAM) stain ×400]. The glomerulus showed mesangial hypercellularity and cellular crescent formation. The basement membrane was thickened, and polymorphonuclear cell infiltration was observed in the tufts. b Light microscopy findings of the second biopsy (PAM stain ×400). Mesangial hypercellularity and polymorphonuclear cell infiltration were still observed. There was no crescent formation in the specimen. c An immunofluorescent study of the C3 antigen in the first biopsy (×400). The C3 deposits were located in the mesangial area and the peripheral capillary walls. d An immunofluorescent study of the nephritis-associated plasmin receptor (NAPlr) antigen in the first biopsy (×400). The NAPlr antigen was located in the mesangial area, showing a granular and ring-like pattern. e An immunofluorescent study of the NAPlr antigen in the second biopsy specimen (×400). The NAPlr antigen was not detected in the glomeruli. f Electron microscopy findings of the second renal biopsy (×6,000). Electron-dense deposits were detected mainly in the mesangial area. A polymorphonuclear cell infiltrated the capillary of the glomerulus
The patient’s clinical course is shown in Fig. 2. Serum ASO titer was elevated immediately after the onset of the symptoms, and it gradually decreased thereafter. As this case exhibited nephrotic syndrome and highly active glomerular lesions with crescent formations, prednisolone was started orally at a dose of 40 mg daily. The patient’s blood pressure was controlled to under 130/80 mmHg using an ARB and a calcium-channel blocker. Following these treatments, urinary protein excretion gradually decreased but hypocomplementemia persisted. A second biopsy was therefore performed, after obtaining the patient’s informed consent, 6 months after initiation of steroid therapy.

Patient’s clinical course. After prednisolone was started orally, the UPE gradually decreased, whereas the hypocomplementemia persisted throughout the observation period. As the corticosteroid therapy could not provide clinical remission and histological improvement, cyclosporine was administered 9 months after initial diagnosis. Serum ASO titer was elevated immediately after symptom onset and gradually decreased thereafter. A second rise in the serum ASO titer, which was observed several months after the initial symptoms, was followed by a decrease in serum C3 levels. In this case, renal biopsies were performed twice at intervals of 6 months for evaluation. ASO antistreptolysin O antibody, UPE urinary protein excretion, RBx renal biopsy
Renal tissue from the second biopsy contained a total of 24 glomeruli, four of which showed global glomerular sclerosis. The remaining glomeruli still showed hypercellularity, but cellular or fibrocellular crescents were not detected (Fig. 1b). Fluorescent immunostaining for C3 was faintly positive in the mesangial area and the peripheral capillary walls, but the intensity was obviously decreased compared with that in the first biopsy (data not shown). Furthermore, the NAPlr antigens in the glomeruli had almost completely disappeared (Fig. 1e). In the electron microscopic examination, the electron-dense deposits were noted in the mesangial area and also found to a lesser extent in the subendothelial spaces, subepithelial spaces, and basement membrane (Fig. 1f). Findings of the second biopsy again showed characteristics consistent with MPGN.
As corticosteroid monotherapy did not lead to clinical remission and histological improvement, cyclosporine was administered 4 months after the second biopsy. However, hypocomplementemia (C3 17 mg/dl, C4 11 mg/dl, CH50 17.3 U/ml) and proteinuria (>1 g/day) persisted even 5 months after the combined administration of the steroid and cyclosporine.
Discussion
This case showed prolonged hypocomplementemia and histological findings consistent with MPGN in serial renal biopsies. Of note, the patient had an elevated serum ASO titer and positive immunostaining for NAPlr antigens in the glomeruli of the first biopsy. We finally diagnosed this case as idiopathic MPGN, as the hypocomplementemia persisted for more than 1 year and we found no manifestations of secondary MPGN, except for the possibility of a latent streptococcal infection.
NAPlr antigen has been reported to be detected in glomeruli during the early stages of APSGN, and the staining diminishes within several months [2]. The NAPlr antigen in glomeruli has been reported to be located in mesangial cells, endothelial cells, and neutrophils, which are known to be similar to the localization of streptococcal pyogenic exotoxin B (SPEB) antigen [6]. These temporal and spatial depositions of the NAPlr antigen are fully consistent with those observed in our case. Moreover, the first renal biopsy specimen showed advanced endocapillary proliferation with neutrophils and CD68-positive cells in glomeruli (Fig. 3a, c, e). These findings further supported the possible involvement of a streptococcal infection. Glomerular NAPlr deposition has also been found in patients with glomerular diseases other than APSGN, including those with Henoch–Schönlein nephritis (HSPN), lupus nephritis, and dense-deposit disease [2, 5, 7–9]. In a study of childhood HSPN cases, it was reported that 30% showed immunostaining for the glomerular NAPlr antigen [5]. However, except for the case presented here, no detailed idiopathic MPGN cases with NAPlr deposition have been previously reported.

Kidney biopsy findings. a Light microscopy findings of the first biopsy specimen (HE stain ×400). Glomerulus showed mesangial hypercellularity and endocapillary proliferation. Neutrophils and mononuclear cells were found to infiltrate to the tufts. b Light microscopy findings of the second biopsy specimen (HE stain ×400). Mesangial hypercellularity and endocapillary proliferation were still observed. c Staining for myeloperoxidase in the first biopsy specimen (×400). Many myeloperoxidase-positive cells were observed in the capillary of the glomerulus. d Staining for myeloperoxidase in the second biopsy specimen (×400). Myeloperoxidase-positive cells still existed in tufts of the glomerulus. e Immunostaining for CD68 using the peroxidase–antiperoxidase method in the first renal biopsy (×400). CD68-positive cells were observed in the capillary of the glomerulus. f Immunostaining for CD68 using the peroxidase–antiperoxidase method in the second renal biopsy (×400). CD68-positive cells were still observed. HE hematoxylin and eosin, RBx renal biopsy
Previous reports indicate that 30–40% of idiopathic MPGN cases had an elevated serum ASO titer [10]. It was also demonstrated that HSPN cases with an elevated ASO titer showed higher rates of positive immunostaining for the glomerular NAPlr antigen than those with a low serum ASO titer [5]. In our case, serum ASO titer was elevated immediately after symptom onset and gradually decreased thereafter. These findings are consistent with the serial changes in glomerular NAPlr immunostaining observed in our case. Interestingly, a second increase in serum ASO titer, observed several months after initial symptoms, was likely to have been followed by the decrease in serum C3 levels. Although we did not check the glomerular NAPlr antigen at that time, such an increase indicates the possibility that another streptococcal infection may have additionally influenced glomerular injury. Another possible explanation for changes in ASO levels is corticosteroid dose insufficiency. Serum ASO titer increased as corticosteroids were tapered to <25 mg/day, whereas it tended to decrease after an immunosuppressive drug was added. In fact, the endocapillary proliferation of the glomeruli was still observed in a second renal biopsy, even when serum ASO titer decreased temporarily (Fig. 3b, d, f). Therefore, prolonged elevation of the ASO titer and the findings of glomerular endocapillary proliferation might represent the exaggerated immune response induced by a streptococcal infection. Such an immune response disorder might therefore influence the persistence of hypocomplementemia and the development of MPGN.
As this case was diagnosed as preeclampsia before the onset of nephritic syndrome, it is necessary to mention histological renal changes that are associated with preeclampsia. Glomerular lesions of preeclampsia involve MPGN-like changes, including mesangial hypercellularity, duplication of the glomerular capillary wall, and swelling of endothelial cells. Although these changes typically recover within a few months, some cases have been reported in which such changes persisted as long as 2 years [11]. These histological findings are consistent with those found in our case. However, preeclampsia does not usually show severe hypocomplementemia, as was seen in our case. Thus, it is not likely that preeclampsia was the main cause of the renal injury in our patient, whereas it is possible that preeclampsia modified the renal endothelial damage and contributed to the development of MPGN lesions. In addition, hydralazine was used for a long period to control blood pressure. Hydralazine is known to induce lupus nephritis or antineutrophil cytoplasmic antibody (ANCA)-related glomerulonephritis [12–14]. However, it is not likely that such hydralazine-related renal injuries underlie the pathology in this case, because neither the clinical nor the histopathological evidence support such etiologies.
In conclusion, we describe a case of idiopathic MPGN with transient glomerular deposition of NAPlr antigen. Further information on cases with similar findings is required to establish the potential roles of streptococcal infections in the development of glomerular diseases of unknown etiology, including idiopathic MPGN.
References
Alchi B, Jayne D. Membranoproliferative glomerulonephritis. Pediatr Nephrol. 2010;25:1409–18.
Yamakami K, Yoshizawa N, Wakabayashi K, Takeuchi A, Tadakuma T, Boyle MD. The potential role for nephritis-associated plasmin receptor in acute poststreptococcal glomerulonephritis. Methods. 2000;21:185–97.
Yoshizawa N, Yamakami K, Fujino M, Oda T, Tamura K, Matsumoto K, et al. Nephritis-associated plasmin receptor and acute poststreptococcal glomerulonephritis: characterization of the antigen and associated immune response. J Am Soc Nephrol. 2004;15:1785–93.
Oda T, Yamakami K, Omasu F, Suzuki S, Miura S, Sugisaki T, et al. Glomerular plasmin-like activity in relation to nephritis-associated plasmin receptor in acute poststreptococcal glomerulonephritis. J Am Soc Nephrol. 2005;16:247–54.
Masuda M, Nakanishi K, Yoshizawa N, Iijima K, Yoshikawa N. Group A streptococcal antigen in the glomeruli of children with Henoch-Schönlein nephritis. Am J Kidney Dis. 2003;41:366–70.
Oda T, Yoshizawa N, Yamakami K, Tamura K, Kuroki A, Sugisaki T, et al. Localization of nephritis-associated plasmin receptor in acute poststreptococcal glomerulonephritis. Hum Pathol. 2010;41:1276–85.
Suga K, Kondo S, Matsuura S, Kinoshita Y, Kitano E, Hatanaka M, et al. A case of dense deposit disease associated with a group A streptococcal infection without the involvement of C3NeF or complement factor H deficiency. Pediatr Nephrol. 2010;25:1547–50.
Sawanobori E, Umino A, Kanai H, Matsushita K, Iwasa S, Kitamura H, et al. A prolonged course of Group A streptococcus-associated nephritis: a mild case of dense deposit disease (DDD)? Clin Nephrol. 2009;71:703–7.
Kikuchi Y, Yoshizawa N, Oda T, Imakiire T, Suzuki S, Miura S. Streptococcal origin of a case of Henoch-Schoenlein purpura nephritis. Clin Nephrol. 2006;65:124–8.
Cameron JS, Glasgow EF, Ogg CS, White RH. Membranoproliferative glomerulonephritis and persistent hypocomplementaemia. Br Med J. 1970;4:7–14.
Oe PL, Ooms EC, Uttendorfsky OT, Stolte LA, van Delden L, Graaff P. Postpartum resolution of glomerular changes in edema-proteinuria-hypertension gestosis. Ren Physiol. 1980;3:375–9.
Schoonen WM, Thomas SL, Somers EC, Smeeth L, Kim J, Evans S, et al. Do selected drugs increase the risk of lupus? A matched case-control study. Br J Clin Pharmacol. 2010;70:588–96.
Sarzi-Puttini P, Atzeni F, Capsoni F, Lubrano E, Doria A. Drug-induced lupus erythematosus. Autoimmunity. 2005;38:507–18.
Dobre M, Wish J, Negrea L. Hydralazine-induced ANCA-positive pauci-immune glomerulonephritis: a case report and literature review. Ren Fail. 2009;31:745–8.
Conflict of interest
The authors declare that no conflict of interest exists.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10157_2011_570_MOESM1_ESM.ppt
Figure S1. Electron microscopy findings of the first renal biopsy (x2,500). Electron-dense deposits were detected mainly in the mesangial area. The glomerulus was highly impaired, and the tufts were occupied by infiltrating cells. In addition, mesangial interposition-like structures were observed (white arrow). (PPT 807 kb)
About this article
Cite this article
Okabe, M., Tsuboi, N., Yokoo, T. et al. A case of idiopathic membranoproliferative glomerulonephritis with a transient glomerular deposition of nephritis-associated plasmin receptor antigen. Clin Exp Nephrol 16, 337–341 (2012). https://doi.org/10.1007/s10157-011-0570-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-011-0570-6