
Abstract
Background
Gastric cancer (GC) is the most common and aggressive cancer of the digestive system and poses a serious threat to human health. Since genes do not work alone, our aim was to elucidate the potential network of mRNAs and noncoding RNAs (ncRNAs) in this study.
Methods
Transcriptome data of GC were obtained from TCGA. R and Perl were used to obtain the differentially expressed RNAs and construct a competing endogenous RNA (ceRNA) regulatory network. To investigate the biological functions of differentially expressed RNAs, loss-of-function and gain-of-function experiments were performed. Real-time PCR (RT-qPCR), western blot analysis, dual-luciferase reporter assays and fluorescence in situ hybridization were conducted to explore the underlying mechanisms of competitive endogenous RNAs (ceRNAs).
Results
Based on TCGA data and bioinformatics analysis, we identified the LINC00163/miR-183/A-Kinase Anchoring Protein 12 (AKAP12) axis. We observed that AKAP12 was weakly expressed in GC and suppressed invasion and metastasis in GC cells, which could be abolished by miR-183. In addition, LINC00163 can be used as a ceRNA to inhibit the expression of miR-183, thus enhancing the anticancer effect of AKAP12.
Conclusion
Our results demonstrated that weak LINC00163 expression in GC can sponge miR-183 to promote AKAP12. We established that the LINC00163/miR-183/AKAP12 axis plays an important role in GC invasion and metastasis and may be a potential biomarker and target for GC treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) is a common malignant cancer of the digestive system and poses a serious threat to human health [1]. Although progress has been made in the current knowledge and scientific advances have been made in the diagnosis and treatment of this disease, the long-term survival of GC is still dismal due to the high recurrence rate and distal metastasis rate [2]. Therefore, exploration of new molecular mechanisms and biomarkers to develop GC treatment strategies is needed. Studies have shown that the occurrence and development of GC are related to the destruction of the gene regulatory network. With the completion of the Human Genome Project (HGP) and the parallel development of next-generation sequencing (NGS), human transcriptome analysis has revealed that more than 98% of the transcriptional output encodes noncoding RNAs (ncRNAs). Studies have indicated that the dysregulation of ncRNAs plays a crucial role in regulating oncogenes or tumor suppressor genes [3]. The complex regulatory network between mRNAs and ncRNAs may be the potential cause of the malignant phenotype of GC [4].
Among numerous ncRNAs, long ncRNAs (lncRNAs) are defined as RNAs > 200 nucleotides in length. Previous reports have shown that lncRNAs are involved in many cellular processes, including epigenetics, transcription and post-transcriptional regulation [5]. As “scaffolds”, lncRNAs induce the interaction of proteins; as “guides”, they prompt the combination of protein and genes; as “enhancers”, they regulate transcription of adjacent genes; and as “decoys”, they bind to miRNAs or proteins [6, 7]. Although studies on the involvement of lncRNAs in tumor progression have made great progress, the regulatory mechanism of a large number of lncRNAs on tumors is still unclear. Therefore, it is of great value and significance to understand the role of lncRNAs in tumorigenesis. One theory suggests that lncRNAs can serve as competitive endogenous RNAs (ceRNAs), which can competitively bind miRNAs targeting mRNAs to regulate the expression of mRNAs [8]. So far, the significance of the regulatory mechanism of ceRNA has been reported in melanoma [9], esophageal squamous cell carcinoma [10], breast cancer [11] and other cancers. In the present study, we obtained normal and tumor samples of GC from The Cancer Genome Atlas (TCGA) database. The differentially expressed RNAs were detected by the R package. Then, we built a ceRNA network with Perl language. Subsequently, we focused on the LINC00163/miR-183/A-Kinase Anchoring Protein 12 (AKAP12) axis and explored its role in the malignant phenotype of GC.
Materials and methods
Data download and processing
The RNA sequencing dataset and corresponding clinical records of GC were downloaded from TCGA (TCGA-STAD, https://portal.gdc.cancer.gov/). The raw RNA expression profiles were downloaded using the RTCGA Toolbox package in the R platform. A total of 406 samples included 375 GC tissues and 31 normal tissue samples.
RNA and miRNA sequence data were obtained from the Illumina HiSeq_RNASeq and Illumina HiSeq_miRNASeq sequencing platforms, respectively. The lncRNA sequences and mRNA sequences were included in the RNA sequences. Perl and R language were used to analyze the RNA data.
Screening for differentially expressed RNAs
The packages “edgeR” and “limma” were used to identify differentially expressed lncRNAs (DELs), differentially expressed miRNAs (DEMis) and differentially expressed mRNAs (DEMs). The intersection of two data points was taken as the “real” differentially expressed RNAs. P < 0.05 and |log2FC|≥ 1 were set as the cutoff criteria. In this study, we mainly found downregulated DELs (dDELs), downregulated DEMs (dDEMs), and upregulated DEMis (uDEMis).
The construction of the ceRNA network
First, we used the miRcode database to match the correspondence between dDELs and uDEMis. Next, the target mRNAs of uDEMis were retrieved by the miRDB, mirTarBase and Target Scan databases. Finally, Cytoscape 3.6.1 software (https://www.cytoscape.org/) was used to construct the ceRNA network.
Tissue samples and cell culture
A total of 100 GC specimens were obtained from patients undergoing surgical resection at the Liaoning Province Cancer Hospital and Institute between 2014 and 2015. Among them, thirty-five GC tissues were equipped with matched non-tumorous adjacent tissues. All 100 GC specimens were used for survival analysis. Patients signed informed consent before surgery. The study was permitted by the Ethics Committee, Liaoning Cancer Hospital and Institute. Supplement Table 1 describes the clinicopathological parameters involved in the analysis.
GES-1 (human normal gastric epithelial cell line), SGC-7901 and BGC-823 (human gastric cancer cell lines) were obtained from China Medical University (Shenyang, China). Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen) at 37 °C under 5% CO2 and1% O2. All experiments were repeated three times independently.
Real-time reverse transcription polymerase chain reaction (real-time PCR)
Total RNA was isolated with TRIzol (Invitrogen) cell separation reagent according to the manufacturer's instructions from cells and tissue. A Promega cDNA core kit (Promega, Madison, WI, USA) was used to generate complementary DNA from 500 ng of total RNA. SYBR Green Master Mix (TaKaRa Bio, Inc., Kusatsu, Japan) was used to perform real-time PCR (LightCycler 480; Roche AG, Basel, Switzerland). Each sample was analyzed three times. U6 was used as a loading control. Fold changes in mRNA expression in different cells were determined by 2−ΔΔCT normalization. Each sample was analyzed in triplicate. The sequence information is listed in Supplement Table 2.
Separation of nuclear and cytoplasmic fractions
The total cellular fractions were divided into nuclear and cytoplasmic fractions by the PARIS kit (AM1921; Thermo Fisher Scientific, Yokohama, Japan) according to the manufacturer's protocol. The kit was used to isolate RNA from the same experimental sample and separated the nuclear and cytoplasmic components before RNA was separated.
Western blot analysis
A 10–15% SDS-polyacrylamide gel and PVDF membranes were used to separate and transfer protein (20 μg) from cells. TBS-Tween buffer (20 mM Tris–HCl, 5% nonfat milk, 150 mM NaCl, and 0.05% Tween-20, pH 7.5) was used to block membranes for 1 h at 21 °C after blotting. Then, the members were incubated with primary antibodies overnight at 4 °C (AKAP12, 1:200, Boster Biological Technology and β-actin, 1:4000, Santa Cruz). Finally, membranes were incubated with secondary antibody (1:5000, Santa Cruz). β-Actin acted as a control. The gray value of the proteins was measured by ImageJ (NIH, Bethesda, MD, USA). Averages of three independent data points are presented as the final result.
Lentivirus vector system, plasmids, and cell transfection
The miR-183 mimics and inhibitor were obtained from GeneChem (Shanghai, China).
Both the full-length human AKAP12 and LINC00163 cDNA sequences were subcloned into the pcDNA 3.1 vector. Following the manufacturer’s protocol, Lipofectamine 2000Reagent (Invitrogen) was used to transfect the GC cells.
Scratch motility and transwell invasion assays
Scratch motility assays were used to evaluate cell migration. GC cells were plated into culture inserts (Ibidi, Regensburg, Germany). After incubation for 24 h, the inserts were removed. An inverted microscope (XDS-100, Shanghai Caikon Optical Instrument Co., Ltd., Shanghai, China) was used to capture the wounded monolayer images at 0 and 24 h post-wounding.
A transwell assay was performed to determine cell invasion. Transwell upper chambers coated with gelatin were used to plate GC cells. The lower chambers were coated with 600 μL FBS (30%, Costar, Lowell, MA, USA). Methanol, hematoxylin and eosin were used to fix and stain cells after incubation for 24 h (Sigma-Aldrich, St. Louis, MO, USA). After removal of the upper chambers, the cells on the surface of the lower chambers were defined as migrated cells, which were counted and captured by microscopy at 100× magnification in five fields. The average cell number per field represented the migrated cells.
Cell Counting Kit-8 proliferation assay
Cell Counting Kit-8 (CCK-8) was performed to evaluate the cellular proliferation capacity according to the manufacturer’s instructions (Dojindo Laboratories, Kumamoto, Japan). Microplate reader determined the optical density at a wavelength of 450 nm (Bio-Rad, Hercules, CA, USA).
Immunofluorescence
Cells were fixed with 4% paraformaldehyde in PBS at room temperature for 10 min and permeabilized by 0.1% Triton-X and 1% BSA in PBS for 30 min at room temperature. DNA staining was performed by 4′,6-diamidino-2-phenylindole (DAPI). Biomarkers were detected by antisense probe hybridization at 37 °C for approximately 16 h. Imaging was performed using an ECLIPSE Ni microscope (Nikon, Japan).
Luciferase reporter assay
Luciferase reporters were generated based on the psiCHECK2 vector (Promega). Both the complete 3′UTR of Homosapiens AKAP12 mRNA, which included the predicted miR-183 binding sites, and the full-length sequences of LINC00163 were PCR amplified and cloned into the psiCHECK2 vector. According to the manufacturer's guidelines, luciferase reporter genes were cotransfected with miR-183 mimics and miR-NC into SGC-7901 cells using Lipofectamine 2000. A Dual-Luciferase Reporter Assay System (Promega) and an Infinite M200 PRO microplate reader (Tecan, Shanghai, China) were used to measure the relative luciferase activity.
Statistical analysis
Statistical analysis was performed by the Statistical Package for the Social Sciences (SPSS; IBM, Armonk, NY, USA) version 23.0. All data are presented as the mean ± SD (standard deviation). The comparisons of data were conducted by Student’s t test when homogeneity of variance was satisfied between groups; otherwise, the Wilcoxon-signed rank test was used. Multiple groups were compared using one-way analysis of variance (ANOVA). P < 0.05 was considered statistically significant.
Results
Differentially expressed RNAs in GC
The TCGA-STAD RNA sequence dataset includes 375 GC samples and 31 paracarcinoma tissues. EdgeR and limma were applied to identify the DELs, DEMis and DEMs (fold change ≥ 2, P < 0.05). For edgeR, 3339 uDELs and 925 dDELs (Fig. 1a, d, g), 148 uDEMis and 65 dDEMis (Fig. 1b, e, h), and 3232 uDEMs and 2401 dDEMs (Fig. 1c, f, i) were identified. For limma, 1314 uDELs and 837 dDELs (Sup Fig. 1a, d, g), 59 uDEMis and 67 dDEMis (Sup Fig. 1b, e, h), and 2271 uDEMs and 2208 dDEMs (Sup Fig. 1c, f, i) were identified. Principal component analysis (PCA) was used to evaluate the screening (Fig. 1g–i, Sup Fig. 1g–i). Venny showed the intersection of dDELs (Fig. 1j), uDELs (Sup Fig. 1J), uDEMis (Fig. 1k), dDEMis (Sup Fig. 1k), dDEMs (Fig. 1l), and uDEMs (Sup Fig. 1l). In the present study, we mainly focused on the dDELs, uDEMis and dDEMs. Therefore, 559 dDELs, 44 uDEMis and 1639 dDEMs were included in the follow-up study.

Analysis of differentially expressed lncRNAs between GC and tumor-adjacent normal tissues. a–c Cluster analysis of differentially expressed RNAs between GC and tumor-adjacent normal tissues was done by edgeR. Heatmaps showed the top 50 differentially expressed lncRNAs (DELs), miRNAs (DEMis) and mRNAs (DEMs). d–f The volcano plots showed the distribution of DELs, DEMis and DEMs screened by edgeR. g–i Principal component analysis (PCA) evaluates the screening of DELs, DEMis and DEMs by edgeR. j 559 downregulated DELs were identified by limma and edgeR. k 44 upregulated DEMis were identified by limma and edgeR. l 1639 downregulated DEMs were identified by limma and edgeR
The construction of the ceRNA regulatory network
We used the miRcode database to match the dDELs and uDEMis. A total of 44 correlations between dDELs and uDEMis were found, with 27 dDELs targeting 5 uDEMis (Table 1). Next, based on the miRTarBase, miRDB, and TargetScan databases, we conducted target predictions for the 5 uDEMis. The results revealed 199 DEMi–DEM interactions, and 192 target mRNAs were obtained. These 192 target mRNAs and dDEMs were matched, and 14 target genes were obtained (Fig. 2a, Table 2). Therefore, we obtained 27 dDELs, 5 uDEMis and 14 dDEMs. Cytoscape was used to construct the ceRNA network to show the relationships among DELs, DEMis and uDEMs (Fig. 2b).

The construction of the ceRNA regulatory network. a Venn diagram of dDEMs involved in the ceRNA regulation network. Yellow area: all dDEMs in GC; purple area: target mRNAs of uDEMis; middle area: the dDEMs that are located in both the differential expression and target groups. b The ceRNA regulatory network. Blue: dDELs; green: uDEMis; pink: dDEMs
AKAP12 was identified as a target of miR-183
Based on the above analysis, we believe that the abnormal expression of AKAP12 may be closely related to the malignant phenotype of GC. According to the ceRNA network, miR-183 was a potential regulatory molecule for AKAP12 (Fig. 2b).
The results of western blot and RT-PCR analyses showed that AKAP12 was weakly expressed in BGC-823 and SGC-7901 cells compared with the levels in control GES-1 cells (Fig. 3a, b). In addition, we examined AKAP12 expression in GC and matched non-tumorous adjacent tissues by RT-PCR. Thirty-five matched specimens were included in the study. AKAP12 expression was observed in all GC tissue samples and was downregulated in GC samples compared to that in adjacent noncancerous samples (Fig. 3c). To further verify the clinical significance of AKAP12, another 65 GC specimens were included in the survival analysis. Not surprisingly, the weakly expressed ANGPT2 contributed to the poor survival of the 100 GC patients in both disease-free survival (DFS, X2 = 10.809, P < 0.001, Fig. 3d) and overall survival (OS, X2 = 10.756, P < 0.001, Fig. 3e).

AKAP12 is weakened in GC and identified as a target of miR-183. a, b The relative expression of ANGPT2 in GES-1 and BGC-823 and SGC-7901 cell lines was determined by western blots and RT-PCR. c Compared differences in the expression of AKAP12 between GC and matched tumor-adjacent tissues. Kaplan–Meier curves for DFS (d) and OS (e) of 100 GC patients with low vs. high expression of AKAP12. The median AKAP12 expression was used as the cutoff value. RT-PCR evaluated the expression of miR-183 in GC cells vs GES-1 (f) and GC tissues vs matched adjacent non-tumor tissue (g). h The correlation between AKAP12 transcriptional levels and miR-183 transcriptional levels was measured in the same set of patients by Spearman correlation analysis. i RT-PCR analyzed miR-183 expression in GC cells treated with mimics and inhibitor of miR-183. j Predicted binding sites for miR-183 on the AKAP12 transcript. k, l Western blots and RT-RCR were used to evaluate the expression of AKAP12 in GC cells after transfecting with the mimics of miR-183. m Luciferase activities were measured in SGC-7901 cells cotransfected with a luciferase reporter containing AKAP12 and the mimics of miR-183 or mutant. Data are presented as the relative ratio of Renilla luciferase activity and firefly luciferase activity. Data are shown as the mean ± SD, n = 3. The statistical significance of the data was assessed by Student’s t test. *P < 0.05, **P < 0.01
Furthermore, we detected the expression of miR-183 in GC cells and tissue by RT-PCR. In contrast to AKAP12, miR-183 was overexpressed in BGC-823 and SGC-7901 and GC samples (Fig. 3f, g). AKAP12 expression was negatively correlated with AKAP12 expression in thirty-five GC tissue samples (r = − 0.9341, P < 0.001, Fig. 3h). The predicted results from miRTarBase showed that ANGPT2 contained miR-183 targeting sites (positions 945–966, Fig. 3j).To further study the regulatory relationship between miR-183 and AKAP12, we used mimics and inhibitors of miR-183 to regulate the expression of miR-183 (Fig. 3i). The data verified that miR-183 could negatively regulate AKAP12 expression at both the transcriptional (Fig. 3l) and translational levels (Fig. 3k), as shown by RT-PCR and western blots, respectively. Furthermore, the luciferase reporter plasmid psiCHECK2-AKAP12 was transfected into GC cells to test the effects of miR-183 on AKAP12 expression. The luciferase activity of psiCHECK2- AKAP12 was decreased by the overexpression of miR-183 but not miR-183-mut (Fig. 3m). Based on the NGS results and a series of bioinformatics analyses, we identifiedAKAP12 as a follow-up target. We verified that AKAP12 was the target gene of miR-183 by RT-PCR, western blot and luciferase assays.
The biological function of ANGPT2
To examine the biological function of AKAP12 and to verify the effect of miR-183 expression on it, we transfected an AKAP12 vector for overexpression of AKAP12 in GC cells (Sup Fig. 2a, b). The upregulation of AKAP12 induced by stable transfection was rescued by the overexpression of miR-183 in both SCG-7901 and BGC-823 cells (Fig. 4a, b). Using a transwell assay, we found that upregulating AKAP12 expression significantly inhibited the invasion of SCG-7901 and BGC-823 cells. When miR-183 was overexpressed, the inhibition was partially eliminated (Fig. 4c, d). We conducted scratch assays to investigate the effect of AKAP12 and miR-183 on cell invasion. These experiments showed that AKAP12 overexpression could significantly inhibit the invasion of GC cells, while this effect could be partially abolished by the upregulation of miR-183 (Fig. 4e). Some studies [12, 13] had shown that miR-183 might have an effect on GC proliferation capacity. CCK-8 was applied to evaluate the effect of AKAP12 and miR-183 on proliferation. Unfortunately, the overexpression of AKAP12 did not affect the proliferation ability of GC. However, with increased miR-183 expression, the proliferation ability of GC was significantly enhanced, which was consistent with the research results of Li et al.[12] (Sup Fig. 3a, b). It might mean that overexpression of miR-183 could affect the proliferation of GC, but not through AKAP12. It was similar to what Soh et al. [14] found in breast cancer.

AKAP12 impaired the invasive and metastatic capacity of GC cells. a, b The mimics of miR-183 could reverse the upregulation of AKAP12 by the vector-AKAP12, which was determined by western blot. a SGC-7901; b BGC-823. c, d Transwell assays were used to evaluate the involvement of AKAP12 in AKAP12 overexpression and AKAP12 overexpression + miR-183 mimics. c SGC-7901; d BGC-823. e Scratch assays were used to evaluate the involvement of AKAP12 in AKAP12 overexpression and AKAP12 overexpression + miR-183 mimics. Up: SGC-7901; down: BGC-823. Data are shown as the mean ± SD, n = 3. The statistical significance of the data was assessed by Student’s t test. *P < 0.05, **P < 0.01
In addition, as AKAP12 was overexpressed in GES-1, we verified the effect of miR-183 on AKAP12 in GES-1 cells: overexpressed miR-183 inhibited the expression of AKAP12 at both transcription and protein levels (Sup Fig. 4a, b). Moreover, scrape and transwell assays confirmed that overexpressed miR-183 could inhibit the motility of GES-1 cells (Sup Fig. 4c, d).
LINC00163 shows crosstalk with miR-183 through direct binding
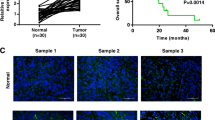
Similar to AKAP12, LINC00163 was also weakly expressed in GC cells compared to GES-1 cells (Fig. 5a, c).The results of nuclear/cytoplasmic RNA fractionation from the subcellular distribution assay showed that LINC00163 was mainly localized in the cytoplasm of GC cells (Fig. 5b, c). Moreover, immunofluorescence and fluorescence microscopy showed the same results (Fig. 5d).

LINC00163 showed crosstalk with miR-183 through direct binding. a RT-PCR exhibited the expression of LINC00163 in GC cells and GES-1. The expression level of LINC00163 in the subcellular fractions of SGC-7901 (b) and BGC-823 (c) cells was detected by RT-PCR. U6 and β-actin were used as nuclear and cytoplasmic markers, respectively. d Immunofluorescence indicates that LINC00163was mainly localized in the cytoplasm. e RT-PCR evaluated the expression of LINC00163 in GC tissues vs matched adjacent non-tumor tissue. f The correlation between LINC00163 transcriptional levels and miR-183 transcriptional levels was measured in the same set of patients by Spearman correlation analysis. g Real-time PCR analyzed LINC00163 expression in GC cells treated with inhibitor of miR-183. h Real-time PCR analyzed miR-183 expression in GC cells with the upregulation of LINC00163. i Luciferase activity in SGC-7901 cells cotransfected with luciferase reporter containing LINC00163and the mimics of miR-183 or mutant. Data are presented as the relative ratio of Renilla luciferase activity and firefly luciferase activity. j Western blot analyzed AKAP12 expression in GC cells with the upregulation of LINC00163. Data are shown as the mean ± SD, n = 3. The statistical significance of the data was assessed by Student’s t test. *P < 0.05, **P < 0.01
Unsurprisingly, LINC00163 expression was similarly weak in GC tissue (Fig. 5e).
In addition, LINC00163 and miR-183 were negatively correlated in GC tissues (Fig. 5f, r = − 0.9341, P < 0.001). To identify whether LINC00163 can interact with miR-183 and act as a ceRNA, we examined the relationship between their expression levels. The expression of LINC00163 was upregulated by the miR-183 inhibitor (Fig. 5g). However, the expression of miR-183, which was originally high in GC cells, was downregulated by the LINC00163 vector (Fig. 5h, Sup Fig. 2c, d). We then transfected the luciferase reporter plasmid psiCHECK2-LINC00163 into GC cells to detect the potential effect of the miRNA on lncRNA expression. Overexpression of miR-183, but not miR-183-mut, decreased the luciferase activity of psiCHECK2-LINC00163 (Fig. 5i). In addition, AKAP12 expression was improved after the overexpression of LINC00163 (Fig. 5j). These data suggested that LINC00163 can directly bind with miR-183 and affect its expression.
LINC00163 acts as a ceRNA to regulate cell invasion
To detect the biological function of LINC00163, we altered its expression. With gain of function of LINC00163 (Sup Fig.2c, d), the expression of AKAP12 was also increased. The upregulation of AKAP12 induced by the LINC00163vector was rescued by induction of miR-183 in both SCG-7901 and BGC-823 cells (Fig. 6a, b). LINC00163 upregulation led to a decreased invasion capacity, as measured by transwell assays. Unsurprisingly, the inhibition of miR-183 partially abolished this effect (Fig. 6c, e). In addition, a scratch assay was used to evaluate the invasive capacity. As expected, the malignant proliferation of GC can be inhibited by high expression of LINC00163, and miR-183 can also partially rescue this biological function (Fig. 6e). As we mentioned above, miR-183 could affect GC proliferation, and we verified the role of LINC00163 in this phenomenon. As expected, the malignant proliferation of GC can be inhibited by the overexpression of LINC00163 and miR-183 could partially rescue this biological function (Sup Fig. 3c, d).

LINC00163 impaired the invasive and metastatic capacity of GC cells. a, b miR-183 mimics reversed the LINC00184-mediated upregulation of AKAP12, as determined by western blot analysis. a SGC-7901; b BGC-823. c, d Transwell assays were used to evaluate the involvement of LINC00163in invasion in the LINC00163-overexpressing and LINC00163-overexpressing + miR-183 mimic groups. c SGC-7901; d BGC-823. e Scratch assays were used to evaluate the involvement of LINC00163in invasion in the LINC00163-overexpressing and LINC00163-overexpressing + miR-183 mimic groups. Up: SGC-7901; down: BGC-823. Data are shown as the mean ± SD, n = 3. The statistical significance of the data was assessed by Student’s t test. *P < 0.05, **P < 0.01
Discussion
With the development of NGS technology and molecular biology, the critical role of lncRNAs in tumorigenesis and development has been confirmed by accumulating lncRNA profiling and functional studies of various types of cancers [15, 16]. LncRNAs are regarded as promising diagnostic biomarkers, attractive therapeutic targets and effective prognostic predictors for GC [17,18,19]. In this study, we obtained transcriptome data for TCGA-STAD. A series of bioinformatics analyses were conducted to explore the regulatory networks of GC-related molecules.
Both “limma” and “edgeR” were applied to analyze differentially expressed RNAs, and the intersection was taken to obtain more accurate data analysis results. Subsequently, based on the negative regulatory relationship in the ceRNA regulatory network, we focused on weakly expressed lncRNAs, mRNAs and overexpressed miRNAs in GC. Finally, we obtained a molecular regulatory network consisting of 27 dDELs, 5 uDEMis and 14 dDEMs. We focused on the LINC00163/miR-183/AKAP12 axis in further studies.
AKAP12, which has three isoforms, was first found in myasthenia gravis and initially identified as an autoantigen [20]. Successive studies have shown that it is lost in a variety of human malignancies, such as prostate cancer [21], gastric cancer [22] and colon cancer [23]. Epigenetic modification, such as promoter hypermethylation or gene deletion, may be the reason for the downregulation of AKAP12 [24]. The low expression of AKAP12 may also be related to the inhibition of Ras, Myc and other oncogenes [25]. In addition, as a scaffold protein, AKAP12 interacted with the PKA and PKC protein kinases (PK), cyclins, F-actin and many other signaling factors to activate the corresponding signaling pathways [14]. Su et al. suggested that AKAP12 suppresses metastatic motility by inhibiting the PKC–Raf/MEK/ERK pathways [26]. Both Regina et al. and Soh et al. suggested that the cell migration of breast cancer was associated with weak expression of AKAP12 [14, 27]. Re-expression of AKAP12 in prostate cancer cells significantly reduced the incidence of lung metastasis [28]. Studies had also shown that AKAP12 was overexpressed in Barrett’s esophagus and esophageal neoplastic and as a molecular biomarker for Barrett’s esophagus diagnosis [29, 30]. Of course, this might be related to the change of the local microenvironment caused by gastroesophageal reflux. In this study, we verified the anticancer effect of AKAP12 in inhibiting GC cell invasion and metastasis through a series of cell function experiments. These findings were consistent with our bioinformatics analysis. However, the regulatory mechanism of AKAP12 in GC remains to be clarified, especially the role of ncRNAs in the regulation of AKAP12. Based on bioinformatics analysis, we predicted that miR-183 was the upstream miRNA that regulates AKAP12. Therefore, we further investigated the biological role of these two RNAs in GC. MiR-183 is located on chromosome 7q32.2 and is involved in the regulation of tumor development as an oncogene [31, 32]. Several published studies have demonstrated that miR-183 expression levels were upregulated in GC compared with their matched adjacent non-tumorous counterparts [12, 31]. In contrast, Xu et al. found that miR-183 was significantly downregulated in GC tissues [13]. Therefore, the carcinogenic role of miR-183 in the development of gastric cancer remains controversial. In the current study, we found that miR-183 was overexpressed in GC cells and tissues and could specifically bind to the 3′untranslated region (UTR) of AKAP12 mRNA, thus inhibiting the expression of AKAP12. In addition, the invasion and metastasis ability of GC were altered by changes in the expression of miR-183 and AKAP12. Rescue experiments proved that the influence of miR-183 on the malignant phenotype of gastric cancer was realized by the expression change of AKAP12. In addition, the experimental results showed that miR-183 can affect the proliferation of GC, but not through AKAP12. A similar biological function of AKAP12 had been reported in breast cancer [14]. Compared with the inhibitory effect of miR-183 on AKAP12, our experimental results showed that there was a positive regulatory relationship between LINC00163 and AKAP12, and gain of function of LINC00163 could inhibit the invasion and metastasis of GC. It suggested that weak expression of LINC00163 was closely related to the malignant phenotype of GC. Similarly, Guo et al. [33] reported LINC00163 was downregulated in lung cancer and negatively correlated with lung cancer metastasis. The low expression of LINC00163 indicates poor prognosis. Similarly, our data also confirmed the anticancer effect of LINC00163, which was weakly expressed in GC and inhibited the invasion and metastasis of GC. Furthermore, miR-183 could also directly bind to LINC00163, affecting its expression and the malignant phenotype of GC. These results lead us to propose a noteworthy regulatory network in which miR-183 and LINC00163 interact to regulate the expression pattern and function of AKAP12.
In recent years, the regulatory role of ncRNAs, especially lncRNAs, at the molecular level has attracted increasing attention. Evidence has shown that lncRNAs can competitively bind miRNAs and mRNAs to regulate the expression levels of mRNAs in cancer [8]. Liu et al. [34] and Song et al. [4] proved that lncRNA HOTAIR, lncRNA-KRTAP5-AS1 and lncRNA-TUBB2A are involved in the malignant phenotype of GC as ceRNAs. The expression of the ZEB family could be regulated by lncRNA-ATB acting as a ceRNA of miR-200 in hepatic cellular carcinoma [35]. An exosome lncRNA, lncARSR, induces sunitinib resistance in renal cancer by endogenous competitive binding of miR-34a and miR-449 [36]. We found that LINC00163 was a tumor suppressor and could competitively bind with miR-183 and affect the expression of AKAP12, thus inhibiting the invasion and metastasis of GC.
Although we revealed and verified the importance of LINC00163/miR-183/AKAP12 in the invasion and metastasis of GC using comprehensive bioinformatics technology and molecular biological approaches, the present study had some limitations. First, the expression and function of LINC00163/miR-183/AKAP12 axis in GC should be validated in in vivo experiments. Second, the mechanism of this axis involved in the signaling pathway still needs to be explained in subsequent studies.
Conclusions
In summary, no gene works alone, and thus, “networks” are important. We identified a LINC00163/miR-183/AKAP12 regulatory network in GC. This molecular mechanism is reported for the first time. AKAP12 inhibited GC cell invasion and metastasis, while miR-183 partially reversed this process. In addition, LINC00163 can affect these functions of AKAP12 as a ceRNA. These results suggest that LINC00163/miR-183/AKAP12 plays an important role in the invasion and metastasis of GC.
References
Siegel RL, Miller KD (2018) Jemal A (2018) Cancer statistics. CA Cancer J Clin 68(1):7–30. https://doi.org/10.3322/caac.21442
Al-Batran SE, Homann N, Pauligk C et al (2019) Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, phase 2/3 trial. Lancet (London, England) 393(10184):1948–1957. https://doi.org/10.1016/s0140-6736(18)32557-1
Schmitt AM, Chang HY (2016) Long noncoding RNAs in cancer pathways. Cancer Cell 29(4):452–463. https://doi.org/10.1016/j.ccell.2016.03.010
Song YX, Sun JX, Zhao JH et al (2017) Non-coding RNAs participate in the regulatory network of CLDN4 via ceRNA mediated miRNA evasion. Nat Commun 8(1):289. https://doi.org/10.1038/s41467-017-00304-1
Bartonicek N, Maag JL, Dinger ME (2016) Long noncoding RNAs in cancer: mechanisms of action and technological advancements. Mol Cancer 15(1):43. https://doi.org/10.1186/s12943-016-0530-6
Morris KV, Mattick JS (2014) The rise of regulatory RNA. Nat Rev Genet 15(6):423–437. https://doi.org/10.1038/nrg3722
Mattick JS, Rinn JL (2015) Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol 22(1):5–7. https://doi.org/10.1038/nsmb.2942
Salmena L, Poliseno L, Tay Y et al (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146(3):353–358. https://doi.org/10.1016/j.cell.2011.07.014
Liu N, Liu Z, Liu X, Chen H (2019) Comprehensive analysis of a competing endogenous RNA network identifies seven-lncRNA signature as a prognostic biomarker for melanoma. Front Oncol 9:935. https://doi.org/10.3389/fonc.2019.00935
Li CY, Zhang WW, Xiang JL et al (2019) Integrated analysis highlights multiple long noncoding RNAs and their potential roles in the progression of human esophageal squamous cell carcinoma. Oncol Rep. https://doi.org/10.3892/or.2019.7377
Wang XW, Guo QQ, Wei Y et al (2019) Construction of a competing endogenous RNA network using differentially expressed lncRNAs, miRNAs and mRNAs in nonsmall cell lung cancer. Oncol Rep. https://doi.org/10.3892/or.2019.7378
Li C, Deng L, Zhi Q et al (2016) MicroRNA-183 functions as an oncogene by regulating PDCD4 in gastric cancer. Anticancer Agents Med Chem 16(4):447–455
Xu L, Li Y, Yan D et al (2014) MicroRNA-183 inhibits gastric cancer proliferation and invasion via directly targeting Bmi-1. Oncol Lett 8(5):2345–2351. https://doi.org/10.3892/ol.2014.2504
Soh RYZ, Lim JP, Samy RP et al (2018) A-kinase anchor protein 12 (AKAP12) inhibits cell migration in breast cancer. Exp Mol Pathol 105(3):364–370. https://doi.org/10.1016/j.yexmp.2018.10.010
Jiang S, Cheng SJ, Ren LC et al (2019) An expanded landscape of human long noncoding RNA. Nucleic Acids Res 47(15):7842–7856. https://doi.org/10.1093/nar/gkz621
Hu Q, Ye Y, Chan LC et al (2019) Oncogenic lncRNA downregulates cancer cell antigen presentation and intrinsic tumor suppression. Nat Immunol 20(7):835–851. https://doi.org/10.1038/s41590-019-0400-7
Zhang E, He X, Zhang C et al (2018) A novel long noncoding RNA HOXC-AS3 mediates tumorigenesis of gastric cancer by binding to YBX1. Genome Biol 19(1):154. https://doi.org/10.1186/s13059-018-1523-0
Zhuo W, Liu Y, Li S et al (2019) Long noncoding RNA GMAN, up-regulated in gastric cancer tissues, is associated with metastasis in patients and promotes translation of ephrin A1 by competitively binding GMAN-AS. Gastroenterology 156(3):676–691.e611. https://doi.org/10.1053/j.gastro.2018.10.054
Sun TT, He J, Liang Q et al (2016) LncRNA GClnc1 promotes gastric carcinogenesis and may act as a modular scaffold of WDR5 and KAT2A complexes to specify the histone modification pattern. Cancer Discov 6(7):784–801. https://doi.org/10.1158/2159-8290.cd-15-0921
Gordon T, Grove B, Loftus JC et al (1992) Molecular cloning and preliminary characterization of a novel cytoplasmic antigen recognized by myasthenia gravis sera. J Clin Investig 90(3):992–999. https://doi.org/10.1172/jci115976
Xia W, Unger P, Miller L et al (2001) The Src-suppressed C kinase substrate, SSeCKS, is a potential metastasis inhibitor in prostate cancer. Cancer Res 61(14):5644–5651
Choi MC, Jong HS, Kim TY et al (2004) AKAP12/Gravin is inactivated by epigenetic mechanism in human gastric carcinoma and shows growth suppressor activity. Oncogene 23(42):7095–7103. https://doi.org/10.1038/sj.onc.1207932
Liu W, Guan M, Su B et al (2010) Quantitative assessment of AKAP12 promoter methylation in colorectal cancer using methylation-sensitive high resolution melting: correlation with Duke's stage. Cancer Biol Ther 9(11):862–871. https://doi.org/10.4161/cbt.9.11.11633
Gelman IH (2010) Emerging roles for SSeCKS/Gravin/AKAP12 in the control of cell proliferation, cancer malignancy, and barrier genesis. Genes Cancer 1(11):1147–1156. https://doi.org/10.1177/1947601910392984
Lin X, Nelson PJ, Frankfort B et al (1995) Isolation and characterization of a novel mitogenic regulatory gene, 322, which is transcriptionally suppressed in cells transformed by src and ras. Mol Cell Biol 15(5):2754–2762. https://doi.org/10.1128/mcb.15.5.2754
Su B, Bu Y, Engelberg D et al (2010) SSeCKS/Gravin/AKAP12 inhibits cancer cell invasiveness and chemotaxis by suppressing a protein kinase C-Raf/MEK/ERK pathway. J Biol Chem 285(7):4578–4586. https://doi.org/10.1074/jbc.M109.073494
Zhou T, Zhang GJ, Zhou H et al (2014) Overexpression of microRNA-183 in human colorectal cancer and its clinical significance. Eur J Gastroenterol Hepatol 26(2):229–233. https://doi.org/10.1097/meg.0000000000000002
Qiu M, Liu L, Chen L et al (2014) microRNA-183 plays as oncogenes by increasing cell proliferation, migration and invasion via targeting protein phosphatase 2A in renal cancer cells. Biochem Biophys Res Commun 452(1):163–169. https://doi.org/10.1016/j.bbrc.2014.08.067
Jin Z, Hamilton JP, Yang J et al (2008) Hypermethylation of the AKAP12 promoter is a biomarker of Barrett's-associated esophageal neoplastic progression. Cancer Epidemiol Prev Biomark 17(1):111
Wang Z, Kambhampati S, Cheng Y et al (2019) Methylation biomarker panel performance in EsophaCap cytology samples for diagnosing Barrett's esophagus: a prospective validation study. Clin Cancer Res 25(7):2127–2135. https://doi.org/10.1158/1078-0432.ccr-18-3696
Tang X, Zheng D, Hu P et al (2014) Glycogen synthase kinase 3 beta inhibits microRNA-183-96-182 cluster via the beta-Catenin/TCF/LEF-1 pathway in gastric cancer cells. Nucleic Acids Res 42(5):2988–2998. https://doi.org/10.1093/nar/gkt1275
Yu BQ, Su LP, Li JF et al (2012) microRNA expression signature of gastric cancer cells relative to normal gastric mucosa. Mol Med Rep 6(4):821–826. https://doi.org/10.3892/mmr.2012.1006
Guo X, Wei Y, Wang Z et al (2018) LncRNA LINC00163 upregulation suppresses lung cancer development though transcriptionally increasing TCF21 expression. Am J Cancer Res 8(12):2494–2506
Liu XH, Sun M, Nie FQ et al (2014) Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer 13:92. https://doi.org/10.1186/1476-4598-13-92
Yuan JH, Yang F, Wang F et al (2014) A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 25(5):666–681. https://doi.org/10.1016/j.ccr.2014.03.010
Qu L, Ding J, Chen C et al (2016) Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell 29(5):653–668. https://doi.org/10.1016/j.ccell.2016.03.004
Funding
Liaoning S&T Project (20180550999), Shenyang young and middle-aged scientific & technological innovation talents support plan (RC180199).
Author information
Authors and Affiliations
Contributions
JZ performed the majority of experiments and analyzed the data and drafted the manuscript; ZZ, YZ designed the research; SG conducted the molecular biology assays and assisted in writing the manuscript; YW collected and analyzed the data; TZ provided critical revision of the manuscript for important intellectual content.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
The study was reviewed and approved by the Faculty of Science Ethics Committee at Liaoning Cancer Hospital and Institute (Cancer Hospital of China Medical University) (20181226).
Informed consent
All patients signed informed consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10147_2019_1604_MOESM2_ESM.pdf
Supplementary file2: Sup Figure 1 Differentially expressed RNAs in GC. A-C. Heatmaps of the expression levels of the top 50 differentially expressed lncRNAs (DELs), miRNAs (DEMis) and mRNAs (DEMs) screened by limma. D-F. Volcano plots of the expression levels of differentially expressed lncRNAs (DELs), miRNAs (DEMis) and mRNAs (DEMs) screened by limma. G-I. Principal component analysis (PCA) evaluates the screening of DELs, DEMis and DEMs by limma. J. The upregulated DELs identified by limma and edgeR. K. The downregulated DEMis identified by limma and edgeR. L. The upregulated DEMs identified by limma and edgeR (PDF 1780 kb)
10147_2019_1604_MOESM3_ESM.pdf
Supplementary file3: Sup Figure 2 A. The vector of AKAP12 upregulated the expression of AKAP12. B. The vector of LINC00163 upregulated the expression of LINC00163 (PDF 374 kb)
10147_2019_1604_MOESM4_ESM.pdf
Supplementary file4: Sup Figure 3 Effect of LINC00163, miR-183 and AKAP12 on cell proliferation. A, B. MiR-183 could promote the proliferation of GC, but AKAP12 had no effect on it. (A. SGC-7901, B. BGC-823). C, D. The inhibition of GC proliferation by LINC00163 could be reversed by overexpressed miR-183. (C. SGC-7901, D. BGC-823) (PDF 520 kb)
10147_2019_1604_MOESM5_ESM.pdf
Supplementary file5: Sup Figure 4 Effect of overexpression of miR-183 on AKAP12 expression and invasion and metastasis in GES-1. A, B. RT-PCR and western blot analyzed AKAP12 expression in GES-1 cells with the upregulated of miR-183. C, D. With the overexpression of miR-183, the migration ability of GES-1 increased (PDF 1986 kb)
About this article

Cite this article
Zhang, J., Piao, Hy., Guo, S. et al. LINC00163 inhibits the invasion and metastasis of gastric cancer cells as a ceRNA by sponging miR-183 to regulate the expression of AKAP12. Int J Clin Oncol 25, 570–583 (2020). https://doi.org/10.1007/s10147-019-01604-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10147-019-01604-w