
Abstract
Small trials have demonstrated promising results utilising intravenous milrinone for the treatment of delayed cerebral ischaemia (DCI) after subarachnoid haemorrhage (SAH). Here we summarise and contextualise the literature and discuss the future directions of intravenous milrinone for DCI. A systematic, pooled analysis of literature was performed in accordance with the PRISMA statement. Methodological rigour was analysed using the MINORS criteria. Extracted data included patient population; treatment protocol; and clinical, radiological, and functional outcome. The primary outcome was clinical resolution of DCI. Eight hundred eighteen patients from 10 single-centre, observational studies were identified. Half (n = 5) of the studies were prospective and all were at high risk of bias. Mean age was 52 years, and females (69%) outnumbered males. There was a similar proportion of low-grade (WFNS 1–2) (49.7%) and high-grade (WFNS 3–5) (50.3%) SAH. Intravenous milrinone was administered to 523/818 (63.9%) participants. Clinical resolution of DCI was achieved in 375/424 (88%), with similar rates demonstrated with intravenous (291/330, 88%) and combined intra-arterial-intravenous (84/94, 89%) therapy. Angiographic response was seen in 165/234 (71%) receiving intravenous milrinone. Hypotension (70/303, 23%) and hypokalaemia (31/287, 11%) were common drug effects. Four cases (0.5%) of drug intolerance occurred. Good functional outcome was achieved in 271/364 (74%) patients. Cerebral infarction attributable to DCI occurred in 47/250 (19%), with lower rates in asymptomatic spasm. Intravenous milrinone is a safe and feasible therapy for DCI. A signal for efficacy is demonstrated in small, low-quality trials. Future research should endeavour to establish the optimal protocol and dose, prior to a phase-3 study.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aneurysmal subarachnoid haemorrhage (SAH) is a neurovascular emergency associated with substantial morbidity and mortality. Delayed cerebral ischaemia (DCI) complicates SAH in the days and weeks that follow, and its prevention and treatment is the cornerstone of SAH management. Treatment of DCI with induced hypertension is now well-established practice, despite a lack of definitive supportive evidence [31, 32]. Earlier institution of intra-arterial spasmolysis is associated with better outcomes [41], but is not without complication risk [2]. Good functional outcome is dependent on the absence of vasospasm and cerebral infarction [72], and improvements in medical therapy would improve patient outcome whilst reducing healthcare costs and interventional risks.
Intravenous milrinone is an emerging therapy for the treatment of DCI, with the potential to address multiple therapeutic targets including macrocirculatory and microcirculatory dysfunction, and vessel inflammation. This systematic review will contextualise and summarise the latest data published on intravenous milrinone for DCI, and highlight potential steps forward for future research.
A primer on the potential effects of milrinone in DCI
Delayed cerebral ischaemia
DCI is the culmination of a complex cascade of neurovascular dysfunction initiated by products of thrombus dissolution (Fig. 1). DCI is defined as the occurrence of focal neurological impairment or a ≥ 2 point decrease in Glasgow Coma Scale, delayed from ictus and aneurysm occlusion, that cannot be attributed to another pathology [95]. Although commonly ascribed to spasm of the large arteries of the Circle of Willis, accumulating evidence suggests other factors may be at play, such as microcirculatory dysfunction, an inverted haemodynamic response to depolarisations, and cortical spreading depressions (of Leão) [23, 66, 67, 89]. In the days following SAH, a complex cascade of interactions between haemoglobin metabolites and vasoactive molecules culminates in a tendency toward vasoconstriction [54]. Key spasmogens include oxyhaemoglobin [73], ferrous iron, adenosine [46, 55], and bilirubin oxidation products [18]. Together, these products of thrombus dissolution are thought to be responsible for both macrocirculatory and microcirculatory dysfunction. Spasm of the large arteries (macrocirculatory dysfunction) is an independent risk factor for DCI, although ischaemic lesions may still occur in its absence [67]. Microcirculatory dysfunction manifests as heterogeneous, and occasionally cessation of, capillary flow [25, 26]. It may be due to disturbed neurovascular coupling, microvascular- and thrombo- inflammation, and an inverted haemodynamic response to cortical spreading depolarisations, the latter possibly mediated by precapillary sphincter dysfunction [34, 48]. Capillary flow heterogeneity reduces oxygen extraction efficiency and predisposes to neuronal calcium overload and hypoxia, culminating in apoptosis/necrosis [44]. Improvement of macrocirculatory and microcirculatory dysfunction is the basis of SAH therapy, as good functional outcome is dependent on the absence of vasospasm and cerebral infarction [72].

Mechanisms underlying DCI. Delayed cerebral ischaemia is a clinical syndrome likely underpinned by a complex interplay of several pathological neurovascular responses. Macrovascular and microvascular dysfunction is initiated by products of thrombus dissolution after SAH. Macrovascular dysfunction can be appreciated radiologically as spasm of the large arteries of the Circle of Willis. Macrovascular spasm is associated with DCI, but whether it is directly involved in its pathogenesis, or rather an epiphenomenon of DCI, is unclear. Cortical spreading depressions are common after SAH, and leave in their wake a prolonged period of cortical electrochemical inhibition. In normal subjects, CSD is generally associated with cerebral hyperaemia. After SAH, however, these periods of cortical inhibition are associated with microvascular dysfunction and cerebral ischaemia. Together, these factors are thought to produce the clinical syndrome of DCI. GCS = Glasgow Coma Scale, rCBF = regional cerebral blood flow, SAH = subarachnoid haemorrhage, DCI = delayed cerebral ischaemia, CSD = cortical spreading depression
Detection of DCI relies on a combination of clinical and radiological assessment [81]. Clinical deterioration should prompt vascular imaging, which may demonstrate angiographic vasospasm or perfusion deficits [20]. The presence of correlated clinical and imaging findings confirms DCI. Discordant findings do not necessarily preclude DCI, as asymptomatic ischaemia and infarction is not uncommon [83], and the limited neurological repertoire of poor-grade patients limits the diagnostic sensitivity of clinical examination [68]. Moreover, current imaging techniques may not detect microvascular or metabolic derangements [15], and thus, DCI in the absence of macrovascular dysfunction may be underdiagnosed.
Pharmacology of milrinone
Milrinone is an inhibitor of the peak III cAMP phosphodiesterase isozyme (PDE3) in cardiomyocytes and vascular smooth muscle. In the heart, it is a positive inotrope, whilst peripherally, it possesses strong vasodilatory effects; milrinone is thus an inodilator. It possesses minimal intrinsic chronotropy and potentiates its action primarily by increasing intracellular cyclic mononucleotide concentration [97]. Milrinone has a half-life of 2.3–2.4 h, a volume of distribution of 0.38 L/kg, is 70% protein bound, and excreted mostly unmetabolised in the urine [99]. As a result, renal dose adjustment may be necessary. Hypokalaemia can occur from intracellular shift, and electrolytes should be regularly monitored during therapy.
Milrinone pharmacodynamics and possible therapeutic mechanisms in DCI
Direct vasodilation
⍺- and β-adrenoceptors are expressed on the endothelium and smooth muscle of the cerebral vasculature [36, 64], with β1-adrenoceptors preferentially upregulated after SAH [93]. The vasodilatory effect of milrinone is due to inhibition of phosphodiesterase and thus increases cAMP and cGMP levels. These cyclic mononucleotides activate cAMP- and cGMP-dependent protein kinases, which through multiple mechanisms, produce vasodilation [39, 60, 74] (Fig. 2). Notably, these pathways are downstream of the adrenoceptor, and thus, the majority of the effect of PDE3 inhibition is independent of the endothelium [47, 50]. Moreover, it is unaffected by changes in adrenoceptor expression or the presence of adrenoceptor antagonists (e.g. “beta-blockers”). Similarly, disruption of the blood-brain barrier does not alter the pial vascular response to milrinone [40]; however, endothelium-dependent relaxation through the NO-cGMP pathway may contribute to vasodilation to a small degree [13].

The effect of milrinone on vascular smooth muscle. B2 andrenergic stimulation, nitrous oxide, and other vasodilatory pathways produce cAMP and cGMP. Phosphodiesterases degrade the phosphodiester bond in cAMP and cGMP producing AMP and GMP, respectively. Inhibition of phosphodiesterase thus increases cAMP and cGMP levels. cAMP and cGMP active cAMP-dependent protein kinase (PKA) and cGMP-dependent protein kinase (PKG), respectively. PKA and PKG have multiple direct effects which induce vasodilation. PKA and PKG increase the open-state probability of the calcium-dependent potassium (KCa) channel, which in turn increases potassium efflux, producing hyperpolarisation. This is augmented by increased frequency of subcellular calcium release, termed “calcium sparks” via ryanodine-sensitive calcium channels of the sarcoplasmic reticulum, which also increase the open-state probability of the KCa channel. Hyperpolarisation closes voltage-dependent calcium channels, leading to reduced intra-cytosolic calcium and thus vasodilation. cAMP and cGMP also directly interact with myosin light chain kinase and phosphatase, leading to dephosphorylation of the myosin light chain and thus vasodilation. AC = adenylyl cyclase, eNOS = endothelial NO synthase, GC = guanylyl cyclase, MLCK = myosin light chain kinase, MLCP = myosin light chain phosphatase, PKA = cAMP-dependent protein kinase, PKG = cGMP-dependent protein kinase
Intra-arterial and intracisternal milrinone dilates cerebral arteries [65] and the effect of intravenous milrinone on cerebral blood flow (CBF) has been reported in small human and non-human animal studies [11, 27, 40, 62, 90]. Overall, milrinone appears to increase CBF; however, these results are limited by the variable methods of measuring CBF and concomitant changes in PCO2, and to a lesser extent, mean arterial pressure (MAP). Increases in CBF from PDE3 inhibition are associated with decreased cerebral vascular resistivity index, increased cardiac output (CO), and potentially an increase in cerebral metabolic rate [62, 63, 82].
Precapillary sphincters lie at the branch points of penetrating arterioles of the brain, and are thought to be at least partly responsible for the prolonged vasoconstriction and oligaemia during cortical spreading depressions [34, 48]. PDE inhibitors have been shown to directly dilate precapillary sphincters [34] and thus may be particularly useful in aborting DCI resulting from cortical spreading ischaemia.
Systemic haemodynamics
Milrinone is a positive inotrope and lusitrope, as well as systemic and pulmonary vasodilator. Increased inotropy and lusitropy are associated with increases in cardiac output (CO) and pulse pressure (Pp), which, combined with vasodilation, decreases mean systemic filling pressure (PMS) and right atrial pressure (RAP). Together, these macrocirculatory effects may improve CBF [16] (Fig. 3).

The effect of milrinone on haemodynamics. Milrinone produces macrocirculatory haemodynamic changes that may theoretically improve CBF. Blue lines demonstrate the effects of milrinone on haemodynamic parameters compared to baseline (shown in black); red arrows highlight the pertinent differences. The left panel demonstrates the increase in CO (or equivalently VR) and decrease in RAP as a result of the inodilator effects of milrinone. A decrease in RAP is transmitted upstream to the venous sinuses. The Davson equation, printed below the left panel, highlights the relationship between venous sinus pressure (Pss) and ICP. Thus, decreased RAP may lead to reductions in ICP and thus improvement in CPP and ultimately increased CBF. The middle panel demonstrates the change in arterial blood pressure pulsatility associated with milrinone therapy, notably an increase in Pp. A wider Pp may induce oscilatory wall shear stress and flow induced vasodilation of the cerebral vasculature, whilst higher systolic peaks may increase flow through collateral vessels, thus increasing CBF. The rightmost panel demonstrates the result of direct vasodilation and endogenous sympatholysis on the cerebral vasculature. The Lassen (pressure-autoregulation) curve is shifted upwards and rightwards, leading to a greater CBF for a given CPP. Together, these mechanisms may explain the indirect effects of milrinone on the cerebral vasculature to increase CBF. VR = venous return, CO = cardiac output, Pp = pulse pressure, Pms = mean systemic filling pressure, RAP = right atrial pressure, CPP = cerebral perfusion pressure, ICPcsf = the component of ICP due to CSF pressure, If = formation rate of CSF, Rout = resistance to CSF outflow, Pss = pressure of the superior sagittal sinus, ABP = arterial blood pressure
Decreased venous pressures may decrease intracranial pressure (ICP), as per the Davson equation [6], increasing cerebral perfusion pressure (CPP). Wall shear stress is the most important regulator of vessel morphology and tone [71]. Increased pulsatility produces oscillatory wall shear stress which may induce NO-mediated dilation of vasospastic territories [45, 92]. High systolic peaks may also open small collateral vessels.
Peripheral baroreceptors are exquisitely sensitive to pulsatile pressure, and sympathetic output increases when Pp decreases independent of MAP [9]. Conversely, increases in Pp and cardiac output produced by milrinone may prompt a reflex sympatholysis of endogenous sympathetic tone. Surgical ablation of sympathetics to the cerebral vasculature is associated with increases in CBF [43, 85, 86, 91], suggesting that reflex sympatholysis would also influence the cerebral circulation. As a result of direct vasodilation as well as downregulation of intrinsic sympathetics, the cerebral autoregulation curve is shifted upwards (i.e. more flow per unit CPP) and the lower limit of autoregulation is shifted rightwards (i.e. maximal vasodilation is achieved at a higher CPP).
Neurogenic cardiomyopathy is common after SAH, subclinical cardiomyopathy perhaps more so, and milrinone therapy may ameliorate failing cardiac haemodynamics in these cases. Finally, the vasodilation induced by milrinone therapy may induce systemic hypotension which compromises CBF to territories with impaired pressure-autoregulation. Indeed, the presence of impaired pressure-autoregulation predicts DCI and poor outcome after SAH [14, 42], and many protocols utilising milrinone employ a concomitant vasopressor.
Endothelial protection and anti-inflammatory properties
Thrombo-inflammation is purported to contribute to microvascular dysfunction after SAH. Products of thrombus dissolution activate leukocytes which secrete pro-thrombotic and pro-inflammatory cytokines. Inflamed endothelium secretes tissue factor (TF) and von Willebrand factor (vWF) which perpetuate microthrombosis. Given the role of vessel inflammation in the pathophysiology of SAH-associated vasospasm, the anti-inflammatory effects of PDE inhibition may contribute to the potential benefit of milrinone therapy in DCI (Fig. 4). PDE3 is highly expressed in monocytes, and milrinone has been shown to reduce both secretion of tumour necrosis factor alpha (TNF-⍺) in models of endotoxaemia [10, 58] and pro-inflammatory interleukins 1β and 6 after cardiopulmonary bypass [37]. PDE inhibition reduces TNF-⍺-dependent nuclear factor-κB reporter gene transcription in vascular smooth muscle, decreasing the expression of inflammatory mediators that facilitate leukocyte chemotaxis and diapedesis such as vascular cell adhesion molecule-1, intercellular adhesion molecule-1, and monocyte chemoattractant protein-1 [4]. Indeed, PDE3 inhibitor–treated mice have significantly reduced numbers of neutrophils and activated microglia invading their brain after experimental stroke [12]. PDE3 inhibition may also have anti-apoptotic effects and neuroprotective effect, with reductions in pro-apoptotic BAX protein and cytochrome C, increases in pro-survival Bcl-2 protein, and reduction of calcium-dependent ischaemic apoptosis seen after treatment in experimental stroke [17, 80].

Endothelial protection and anti-inflammatory properties of milrinone. Microvascular inflammation is an important factor in the pathogenesis and perpetuation of DCI. Products of thrombus dissolution are pro-inflammatory and activate leukocytes to secrete pro-thrombotic and pro-inflammatory cytokines and the endothelium to secrete TF and vWF. This creates a vicious positive feedback loop of microthrombosis and vascular inflammation, which promotes vessel spasm. Milrinone is anti-inflammatory and can downregulate inflammatory signalling in monocytes, breaking this positive feedback loop. PMN = polymorphonuclear cells, TNF-a = tumour necrosis factor alpha, IL = interleukin, ROS = reactive oxygen species, vWF = von Willebrand factor, TF = tissue factor
Methods
A review of the literature was conducted as per the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) statement [57]. The methodological quality of included studies was rated using the Methodological Index for Non-Randomized Studies (MINORS) [87], and was analysed for bias as per the Cochrane Handbook guidelines [38].
Search strategy
The MEDLINE and EMBASE electronic bibliographic databases were consulted for articles written in the English language over the last two decades (January 2000 until May 2020) using the search string ((milrinone OR PDE OR PDE3 OR phosphodiesterase OR olprinone OR levosimendan) AND (vasospasm OR delayed cerebral isch(a)emia OR delayed isch(a)emic neurological deficit OR subarachnoid h(a)emorrhage). The references from included papers were hand-searched for other suitable papers.
Study selection, inclusion, and exclusion criteria
Trials were included if they studied the effects of intravenous milrinone in human patients with aneurysmal subarachnoid haemorrhage experiencing delayed cerebral ischaemia. Studies were excluded if they were single case reports. Where multiple publications utilised the same data set, only the most recent study was included. Studies for which data extraction was not possible were excluded. Studies that only investigated intra-arterial milrinone were excluded, but those that investigated intra-arterial milrinone followed by intravenous milrinone were included (the latter we termed “primary intra-arterial”).
Data extraction
Data on the characteristics of the included studies were extracted including study design; study population including age, gender, and SAH grade; number of participants; milrinone protocol; and haemodynamic monitoring. The primary outcome measure was reversal of neurological deficit attributable to DCI. Secondary outcomes included angiographic response, long-term functional outcome, rate of cerebral infarction attributable to DCI, adverse events, and follow-up duration.
Results
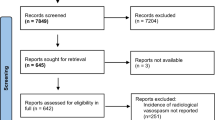
A total of 10 studies were identified from the systematic search of the literature (Fig. 5).

PRISMA diagram of the systemic search process. The search of the electronic databases identified 855 records, which were supplemented by 15 records identified from the bibliographies of studies undergoing full-text review. After removal of duplicates, a total of 565 unique records were screened for relevance, of which 134 underwent full-text review. Of these, 124 were excluded as they were either reviews, case reports, non-human trials, included data that had been subsequently re-published, or did not include intravenous administration of milrinone. A total of 10 studies satisfied our selection criteria
Overview of studies
The 10 studies identified from the literature are summarised in Tables 1, 2, and 3 [1, 5, 7, 21, 22, 29, 51, 61, 78, 94]. All were small or moderate sized cohort studies, either retrospective (n = 5) or prospective (n = 5). Five trials were presented only as conference abstracts. As no studies were randomised controlled trials, as per the Cochrane group, all were of high risk of bias in each domain (selection, performance, detection, attrition, and reporting) [38]. The methodological quality of the studies was analysed using the MINORS criteria [87] (Supplementary Data 1).
A total of 818 patients were enrolled across the 10 studies, with intravenous milrinone administered to 523 (63.9%) participants. The mean age of patients was 52 years, and females (69%) tended to outnumber males, consistent with previous epidemiological studies of subarachnoid haemorrhage [28]. Clinical grading was commonly performed using the World Federation of Neurosurgical Societies (WFNS) system, and across studies that reported individual WFNS grades, there was a similar proportion of low- (WFNS 1–2) (n = 260, 49.7%) and high-grade (WFNS 3–5) (n = 263, 50.3%) SAH.
How milrinone was incorporated into the clinical protocol differed between studies, with the majority using new clinical and/or radiological deterioration as the trigger for initiating therapy. Concurrent vasopressor use was common; however, methodology differed in that some instituted noradrenaline to maintain pre-milrinone baseline blood pressure whilst others sought to hypertense all patients in an effort to improve cerebral perfusion pressures against spastic vessels. Cardiac output and cerebral blood flow were infrequently measured.
Dosing of milrinone varied between studies, and an intravenous bolus prior to commencing infusion was not universal. Boluses, when utilised, ranged from 50 to 200 mcg/kg. Infusion rates were initiated most commonly at 0.75 mcg/kg/min (range 0.15–1.0 mcg/kg/min) with ceiling doses ranging from 0.75 to 2.5 mcg/kg/min (commonly 1.25–1.5 mcg/kg/min). The timing and criteria for increasing the rate of infusion were not reported, nor was the rate of neurological improvement at different infusion rates. Thus, a dose-response relationship could not be established. Primary intra-arterial studies also demonstrated heterogeneous dosing regimens, though 8 mg (range 5–24 mg) was most common.
Reversal of neurological deficit
Five studies investigated the effect of primary intravenous milrinone on neurological deficits associated with DCI [1, 21, 22, 51, 78]. Our primary outcome occurred in 291/330 (88%) of patients receiving primary intravenous therapy [1, 21, 22, 51, 78]. Response rates were similar for primary intra-arterial therapy 84/94 (89%) [5, 7, 21, 29, 94]. Of those that failed to respond to primary intravenous therapy, indications for rescue spasmolysis included new neurological deficit during milrinone therapy (80%), persistent neurological deficit despite intravenous milrinone (10%), and worsening angiographic vasospasm (10%) [1]. Time-dependent efficacy was evident in one small study (n = 40), with 70% of patients experiencing reversal of DCI after 36 h of intravenous therapy, increasing to 100% by 72 h [22].
In a single trial, at the advent of DCI, a vasopressor was instituted to induce hypertension as an adjunct to milrinone [21], whilst in the remaining four, a vasopressor was instituted as necessary to counteract the systemic vasodilatory effect of milrinone and maintain baseline blood pressure. If neurological deficits were refractory to repeat bolus and increased milrinone infusion, these latter protocols called for induced hypertension with escalating doses of vasopressor. No study compared the combination of induced hypertension and milrinone to milrinone and vasopressor as necessary to maintain normotension. Moreover, no study quantified the additional number of patients that achieved reversal of neurological deficits after induced hypertension.
Angiographic outcome
Two studies reported angiographic response to intravenous milrinone. Combined, they demonstrated that 165/234 (71%) patients achieved an angiographic response [21, 22]. Clinically symptomatic vasospasm tended to be more resistant to therapy than vasospasm detected on angiography alone (p = 0.06, our calculation) [21, 22]. Infusion of intravenous milrinone reversed 36 of 70 (51%) vasospastic segments in clinically symptomatic patients and 125 of all 194 vasospastic segments (64%) in one cohort [21]. Results with primary intra-arterial milrinone were similar, reversing 22/35 (63%) vasospastic segments in clinically symptomatic patients and 37/52 (64%) vasospastic segments.
Consistent with previous data, intra-arterial milrinone produced a robust angiographic response [1, 5, 7, 21, 29]. Due to our search strategy, all included studies followed intra-arterial spasmolysis with an intravenous infusion of milrinone. No study compared the effect of spasmolysis alone compared to spasmolysis and subsequent infusion on angiographic or functional outcome.
Haemodynamic outcome
Systemic hypotension (inconsistently defined as SBP < 90 mmHg) was the most common haemodynamic response to intravenous milrinone [1, 21, 22, 29, 51, 78], seen in 70/303 (23%) cases. Tachycardia, either compensatory or a primary effect of milrinone, was reported in five studies [1, 7, 21, 29, 61], although the incidence (9%) was significantly less than with the adrenergic inodilator dobutamine (27%) [61]. Noradrenaline was required in 20–68% of cases when used to supplement milrinone [1, 5, 22, 51, 78]. Moreover, when hypertensive therapy was initiated at the advent of DCI, 45% of cases required an increase of ≥ 25% in vasopressor dose after initiation of milrinone [21].
The effect of intravenous milrinone on non-invasive cerebral oxygen saturation was assessed in a single study [61]. Milrinone was inferior to dobutamine in improving regional cerebral oxygen saturation in vasospastic territories, and this effect appeared dependent on increases in cardiac output [61]. Intra-arterial milrinone significantly improved cerebral blood flow in the corresponding territory [7], consistent with prior reports.
Functional outcome
Functional outcome was reported in nine studies [1, 5, 7, 21, 22, 29, 51, 78, 94], four of which utilised primary intravenous milrinone [1, 21, 22, 51]. Good functional outcome, when defined as modified Rankin Scale (mRS) ≤ 2, was seen in 218/297 (73%) patients treated with primary intravenous milrinone [1, 21, 51]. Similar numbers were seen after primary intra-arterial milrinone, with 67% achieving a mRS ≤ 2 at 12 months post-ictus [21]. In small studies, 92.5% of patients receiving primary intravenous (n = 40) [22] and 80% receiving primary intra-arterial therapy (n = 5) [94] achieved a good outcome when the definition was extended to an mRS ≤ 3.
Infarction related to DCI
Two studies, n = 110 [1] and n = 88 [51], comprised entirely of patients with clinically symptomatic DCI reported vasospasm attributable infarcts in 15% and 36% of patients, respectively. Another study (n = 101), comprised of patients with clinical and/or radiological vasospasm, reported a low overall rate of cerebral infarction (3%), which did not differ between primary intravenous (3%) and primary intra-arterial (4%) therapy [21]. The dissonance between studies suggests that clinically symptomatic vasospasm may be inherently more likely to cause cerebral infarction.
Adverse events
The most common adverse effects of milrinone therapy were hypotension and tachycardia, as discussed above. New or worsening arrhythmia (18/263, 7%) and hypokalaemia (31/287, 11%) were also seen with milrinone therapy; however, their incidence was similar to those who did not receive milrinone [1, 21]. Intolerance to milrinone necessitating downtitration or cessation of the infusion was uncommon, encountered on four instances across all studies.
Each occurred in the setting of a relative contraindication to chronotropic, inodilating agents such as milrinone (including left ventricular outflow tract obstruction and tachyarrhythmia) [1, 21].
Discussion
Key results
Studies utilising intravenous milrinone for the treatment of DCI demonstrate good efficacy in uncontrolled cohorts. Clinical and angiographic vasospasm responded to intravenous milrinone in over two-thirds of cases, with hypotension and tachycardia the most common haemodynamic effects of therapy. Good functional outcome was achieved in the majority of patients. Overall, in a small number of uncontrolled, non-randomised cohorts, intravenous milrinone appears a safe and feasible therapy for DCI.
Limitations and unanswered questions
The major limitation of our review is the size and quality of the included studies. We report 10 studies of which only 3 are full journal articles on primary intravenous therapy. Although half of the studies were prospective, none had an adequate control or were randomised. Although a testament to our broad and thorough search process, many studies were presented as conference abstracts alone and thus provided limited data on safety and secondary outcomes. Findings are subject to selection bias, as patients were not randomised and inclusion criteria for studies varied; reporting bias, as no null studies have been reported; attrition bias, as many trials had patients lost to follow-up; detection bias, as clinicians and outcome assessors were not universally blinded; and may lack external validity, as few centres are experienced with these protocols.
A milrinone-based therapeutic protocol could take many forms (Fig. 6). The most popular framework, the “Montreal protocol” (protocol F, Fig. 6), utilises intravenous milrinone as first-line therapy for DCI, with vasopressors initially only instituted to maintain baseline blood pressure; if DCI is refractory, milrinone is uptitrated and hypertension is induced. A single study demonstrated comparable efficacy between primary intravenous and primary intra-arterial therapy [21]; however, it was not randomised. Whether this finding can be extrapolated to the Montreal protocol is not clear, given the differing utilisation of concurrent vasopressors for hypertensive therapy.

Possible therapeutic protocols involving PDE inhibition. Protocol A depicts standard of care, with induced hypertension for clinical delayed cerebral ischaemia (DCI) after subarachnoid haemorrhage (SAH). Protocol B depicts prophylactic phosphodiesterase inhibitor (PDEi) only, which has been performed previously with intravenous milrinone or oral cilostazol. Protocols C and D depict the addition of intravenous milrinone to standard of care, leading to concurrent hypertensive and PDEi therapy at the onset of DCI, with and without the addition of PDEi prophylaxis. Protocol E represents a more aggressive option, with earlier introduction of intra-arterial therapy, and milrinone used only after spasmolysis. Protocol F depicts what is commonly referred to as the “Montreal Protocol”, first published by the Montreal group, and is similar to protocol C except vasopressors are only used to maintain baseline blood pressure. It is one of the more popular protocols in the literature. Protocol G is similar, but has the addition of prophylactic PDE inhibitor therapy. SAH = subarachnoid haemorrhage; DCI = delayed cerebral ischaemia; IA = intra-arterial; IV = intravenous; PDEi = phosphodiesterase Inhibitor; MAP = mean arterial pressure
The use and size of a bolus dose and the subsequent rate of infusion differed between studies. No study provided data on the efficacy and incidence of side effects at different infusion rates. Further studies, or post hoc analysis of existing data, are thus required to establish the optimal dosing strategy and to confirm a dose-response relationship exists.
Another consideration for future trials is the role of vasopressor support and hypertensive therapy. Induction of hypertension at the onset of clinical DCI is a class 1 recommendation in international guidelines [19], despite a lack of definitive evidence that induced hypertension increases CBF [32] or ameliorates DCI [31] after SAH. HIMALAIA [31], the only randomised controlled trial on induced hypertension, was terminated early due to poor recruitment and lack of effect on CBF in its nested study. Long-term outcomes were not improved by hypertensive therapy, but early termination determined the study was underpowered. Of the non-randomised studies examining the role of induced hypertension on clinical outcome after DCI, vasopressor therapy was generally associated with improved short-term outcomes; however, these failed to translate to any long-term functional benefit [3, 8, 30, 35, 56, 59, 70, 76, 77, 79]. Serious complications, including arrhythmia, haemorrhage, and pulmonary oedema, occurred in up to half of patients, and had a dose-dependent relationship with blood pressure. Several theories have been proposed to reconcile these trial results with the anecdotal clinical benefits of hypertensive therapy. Pressure-autoregulation status may vary between patients and within vascular territories of the same patient, leading some regions or patients to benefit from increased perfusion pressure whilst others do not. Moreover, CBF may not be the optimal surrogate marker of treatment success as induced hypertension may perhaps work at a microcirculatory scale to reduce capillary flow heterogeneity and shunt, providing a more homogenous distribution of blood flow without increasing total CBF. The studies included in this review varied in their utilisation of vasopressor therapy. Whether synchronous vasopressor and inodilator therapy has any detrimental effect on the latter or offers any benefit over either therapy alone remains uncertain, and should be the focus of a future trial (Fig. 7).

Framework for a clinical trial comparing milrinone therapy to hypertensive therapy. Patients with DCI can be randomised to either hypertensive therapy alone, or milrinone infusion with or without concomitant vasopressor. Spasmolysis or angioplasty would be performed for resistant DCI. SAH = subarachnoid haemorrhage; DCI = delayed cerebral ischaemia; IA = intra-arterial; PDEi = phosphodiesterase inhibitor; MAP = mean arterial pressure; BP = blood pressure
Together, we are left with the following unanswered questions:
-
Should milrinone be used prophylactically or reactively?
-
What is the optimal clinical and/or radiological trigger to commence milrinone therapy?
-
What is the optimal dose for intravenous infusion, and does bolus dosing add or detract from efficacy?
-
Should vasopressors be used to provide hypertensive therapy, and if so, should this be an adjunct or rescue therapy in the setting of milrinone infusion?
-
If vasopressors are used only to maintain baseline blood pressure, what degree of blood pressure drop should trigger vasopressor support?
-
What is the optimal trigger of rescue spasmolysis or angioplasty in the setting of milrinone therapy?
-
Are other inodilators or PDE inhibitors superior to milrinone for this purpose?
Generalisability
Whilst milrinone is a widely available, off-patent medication, the expertise and experience required to manage patients with severe DCI with this novel therapy is not broadly available, limiting the generalisability of these results. Moreover, the cardiac and haemodynamic monitoring required during milrinone infusion necessitates a skilled neurocritical care setting. Conversely, in centres where emergent rescue spasmolysis is not always accessible, intravenous milrinone represents a readily implementable therapy for resistant vasospasm.
Future directions
DCI is the clinical endpoint of a complex cascade of circulatory and neuronal dysfunction. Unfortunately, limitations in the pathophysiological understanding of DCI have hindered the development of tailored therapeutic interventions. Even the mechanisms of established therapies (e.g. nimodipine) remain elusive [52]. We hope that with improved understanding of the physiological derangements underlying DCI, in concert with more sophisticated monitoring and imaging, the roles for various therapies in DCI will become clear.
DCI prophylaxis
Prophylactic use of milrinone has been explored in a two studies [33, 88]. Prophylactic milrinone, in addition to induced hypertension with noradrenaline, significantly improved cerebral oxygen saturation compared to induced hypertension alone; however, MAP was significantly greater in the milrinone group [33]. Conversely, when compared to a 500-mg/day infusion of magnesium sulphate, a 0.5-mcg/kg/min infusion of milrinone was associated with a higher incidence of vasospasm, lower GCS scores, and more frequent need for supplemental vasopressors [88]. Cilostazol, an oral PDE3 inhibitor, has been shown to improve outcome when used for DCI prophylaxis [75, 84]. The role of PDE3 inhibitor prophylaxis is an area of ongoing research, and should be considered in future trials assessing therapy for DCI.
Alternative inodilators
Although milrinone appears the favoured agent in the literature, its efficacy compared to other PDE inhibitors and inodilators has not been established robustly. Intravenous sildenafil (a PDE5 inhibitor) improved angiographic vasospasm in 8/12 (67%) cases in one study [96], but failed to demonstrate a consistent improvement in CBF [24]. Alternative inodilators such as levosimendan, a PDE3 inhibitor [69], and dobutamine, a β-adrenoceptor agonist, have also been proposed as therapies for DCI. In a series of 5 patients with SAH, dobutamine was shown to increase CO and CBF independent of changes to MAP [49] and dobutamine has also been shown to improve neurological deficits associated with DCI [53] and improve cerebral oxygenation [61] after SAH. Olprinone, another PDE3 inhibitor, has been shown to increase cerebral blood flow in patients with SAH [82], stroke, and healthy controls [98]. Whether all inodilators and PDE inhibitors are as efficacious, or whether milrinone or another agent possesses intrinsic properties that confer it, superiority requires further investigation.
Conclusion
The literature presented in this review demonstrates that milrinone is safe, feasible, and possibly highly effective. Milrinone achieved clinical and angiographic response in over two-thirds of cases, and thus may decrease the need for invasive rescue spasmolysis and its associated risks. Although a signal for efficacy has been demonstrated, the literature is limited and overall of low quality. Further research, with rigorous methodology, is required to confirm these findings. The role of induced hypertension and vasopressor support, the role of prophylactic PDE inhibitor therapy, and the optimal trigger for initiation of milrinone and rescue therapy also require further investigation. Determining if and how milrinone exerts a protective effect will be crucial to establishing its specific role in the treatment of DCI. The ability to identify specific derangements (large artery spasm, microcirculatory dysfunction, cortical spreading ischaemia) in each patient and provide tailored therapy for each contributory pathology is an exciting prospect.
Data Availability
All data generated or analysed during this study are included in this published article (and its supplementary information files).
References
Abulhasan YB, Ortiz Jimenez J, Teitelbaum J, Simoneau G, Angle MR (2020) Milrinone for refractory cerebral vasospasm with delayed cerebral ischemia. J Neurosurg 1:1–12. https://doi.org/10.3171/2020.1.JNS193107
Adami D, Berkefeld J, Platz J, Konczalla J, Pfeilschifter W, Weidauer S, Wagner M (2019) Complication rate of intraarterial treatment of severe cerebral vasospasm after subarachnoid hemorrhage with nimodipine and percutaneous transluminal balloon angioplasty: Worth the risk? J Neuroradiol J Neuroradiol 46:15–24. https://doi.org/10.1016/j.neurad.2018.04.001
Aiyagari V, Cross DT, Deibert E, Dacey RG, Diringer MN (2001) Safety of hemodynamic augmentation in patients treated with Guglielmi detachable coils after acute aneurysmal subarachnoid hemorrhage. Stroke 32:1994–1997. https://doi.org/10.1161/hs0901.094621
Aizawa T, Wei H, Miano JM, Abe J, Berk BC, Yan C (2003) Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ Res 93:406–413. https://doi.org/10.1161/01.RES.0000091074.33584.F0
Alamri AS, A A, D T, M A, B L, M L, M B, J T (2016) Use of intra-arterial milrinone rescue therapy in patients with refractory and super refractory vasospasm after aneurysmal subarachnoid hemorrhage. J Neurol Sci. https://doi.org/10.1017/cjn.2016.87
Andersson N, Malm J, Eklund A (2008) Dependency of cerebrospinal fluid outflow resistance on intracranial pressure: clinical article. J Neurosurg 109:918–922. https://doi.org/10.3171/JNS/2008/109/11/0918
Arakawa Y, Kikuta K, Hojo M, Goto Y, Ishii A, Yamagata S (2001) Milrinone for the treatment of cerebral vasospasm after subarachnoid hemorrhage: report of seven cases. Neurosurgery 48:723-8-discussion 728-30. https://doi.org/10.1097/00006123-200104000-00004
Awad IA, Carter LP, Spetzler RF, Medina M, Williams FC (1987) Clinical vasospasm after subarachnoid hemorrhage: response to hypervolemic hemodilution and arterial hypertension. Stroke 18:365–372. https://doi.org/10.1161/01.str.18.2.365
Barrett KE, Barman SM, Boitano S, Brooks H (2015) Ganong’s Review of Medical Physiology, 25th edn. McGraw Hill Professional
Bergman MR, Holycross BJ (1996) Pharmacological modulation of myocardial tumor necrosis factor alpha production by phosphodiesterase inhibitors. J Pharmacol Exp Ther 279:247–254
Bianchi MO, Cheung P-Y, Phillipos E, Aranha-Netto A, Joynt C (2015) The effect of milrinone on splanchnic and cerebral perfusion in infants with congenital heart disease prior to surgery. Shock (Augusta, Ga) 44:115–120. https://doi.org/10.1097/shk.0000000000000388
Bieber M, Schuhmann MK, Volz J, Kumar GJ, Vaidya JR, Nieswandt B, Pham M, Stoll G, Kleinschnitz C, Kraft P (2019) Description of a novel phosphodiesterase (PDE)-3 inhibitor protecting mice from ischemic stroke independent from platelet function. Stroke 50:478–486. https://doi.org/10.1161/STROKEAHA.118.023664
Birk S, Edvinsson L, Olesen J, Kruuse C (2004) Analysis of the effects of phosphodiesterase type 3 and 4 inhibitors in cerebral arteries. Eur J Pharmacol 489:93–100. https://doi.org/10.1016/j.ejphar.2004.02.038
Budohoski KP, Czosnyka M, Kirkpatrick PJ, Smielewski P, Steiner LA, Pickard JD (2013) Clinical relevance of cerebral autoregulation following subarachnoid haemorrhage. Nat Rev Neurol 9:152–163. https://doi.org/10.1038/nrneurol.2013.11
Castle-Kirszbaum M, Ayton S, Goldschlager T (2020) Letter to the Editor. Hyperglycolysis as a common cause for elevated lactate in subarachnoid hemorrhage. J Neurosurg 1–2. https://doi.org/10.3171/2020.1.JNS20191
Castle-Kirszbaum M, Parkin WG, Goldschlager T, Lewis PM (2021) Cardiac output and cerebral blood flow: a systematic review of cardio-cerebral coupling. J Neurosurg Anesthesiol. In Press 1–33
Choi JM, Shin HK, Kim KY, Lee JH, Hong KW (2002) Neuroprotective effect of cilostazol against focal cerebral ischemia via antiapoptotic action in rats. J Pharmacol Exp Ther 300:787–793. https://doi.org/10.1124/jpet.300.3.787
Clark JF, Reilly M, Sharp FR (2002) Oxidation of bilirubin produces compounds that cause prolonged vasospasm of rat cerebral vessels: a contributor to subarachnoid hemorrhage-induced vasospasm. J Cereb Blood Flow Metab 22:472–478. https://doi.org/10.1097/00004647-200204000-00011
Sander CE, Rabinstein AA, Ricardo CJ, Derdeyn CP, Jacques D, Higashida RT, Hoh BL, Kirkness CJ, Naidech AM, Ogilvy CS, Patel AB, Gregory TB, Paul V (2012) Guidelines for the management of aneurysmal subarachnoid hemorrhage. Stroke 43:1711–1737. https://doi.org/10.1161/STR.0b013e3182587839
Cremers CHP, van der Schaaf IC, Wensink E, Greving JP, Rinkel GJE, Velthuis BK, Vergouwen MDI (2014) CT perfusion and delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. J Cereb Blood Flow Metab 34:200–207
Crespy T, Heintzelmann M, Chiron C, Vinclair M, Tahon F, Francony G, Payen J-F (2019) Which protocol for milrinone to treat cerebral vasospasm associated with subarachnoid hemorrhage? J Neurosurg Anesthesiol 31:323–329. https://doi.org/10.1097/ana.0000000000000527
De Leon A, Polderman K, Abrego G c, Alfaro F (2014) Milrinone to improve outcome in cerebral vasospasm after SAH. From april 2012 to april 2013 neuro-ICU caja del seguro social. Panama. Neurocritical Care. https://doi.org/10.1007/s12028-014-0034-4
Dhar R, Diringer MN (2014) Relationship between angiographic vasospasm, cerebral blood flow, and cerebral infarction after subarachnoid hemorrhage. Springer International Publishing, pp 161–165
Dhar R, Washington C, Diringer M, Zazulia A, Jafri H, Derdeyn C, Zipfel G (2016) Acute effect of intravenous sildenafil on cerebral blood flow in patients with vasospasm after subarachnoid hemorrhage. Neurocrit Care 25:201–204. https://doi.org/10.1007/s12028-016-0243-0
Dreier JP (2011) The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17:439–447. https://doi.org/10.1038/nm.2333
Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, Tolias C, Oliveira-Ferreira AI, Fabricius M, Hartings JA, Vajkoczy P, Lauritzen M, Dirnagl U, Bohner G, Strong AJ, group C study (2009) Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 132:1866–1881. https://doi.org/10.1093/brain/awp102
Drexler H, Höing S, Faude F, Wollschläger H, Just H (1987) Central and regional vascular hemodynamics following intravenous milrinone in the conscious rat: comparison with dobutamine. J Cardiovasc Pharmacol 9:563–569. https://doi.org/10.1097/00005344-198705000-00010
Eden SV, Meurer WJ, Sánchez BN, Lisabeth LD, Smith MA, Brown DL, Morgenstern LB (2008) Gender and ethnic differences in subarachnoid hemorrhage. Neurology 71:731–735. https://doi.org/10.1212/01.wnl.0000319690.82357.44
Fraticelli AT, Cholley BP, Losser M-R, Saint Maurice J-P, Payen D (2008) Milrinone for the treatment of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke 39:893–898. https://doi.org/10.1161/STROKEAHA.107.492447
Frontera JA, Fernandez A, Schmidt JM, Claassen J, Wartenberg KE, Badjatia N, Connolly ES, Mayer SA (2010) Clinical response to hypertensive hypervolemic therapy and outcome after subarachnoid hemorrhage. Neurosurgery 66:35–41. https://doi.org/10.1227/01.neu.0000359530.04529.07
Gathier CS, van den Bergh WM, van der Jagt M, Verweij BH, Dankbaar JW, Müller MC, Oldenbeuving AW, Rinkel GJE, Slooter AJC, Group H-S (2018) Induced hypertension for delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: a randomized clinical trial. Stroke 49:76–83. https://doi.org/10.1161/strokeaha.117.017956
Gathier CS, Dankbaar JW, van der Jagt M, Verweij BH, Oldenbeuving AW, Rinkel GJE, van den Bergh WM, Slooter AJC, Group H-S (2015) Effects of induced hypertension on cerebral perfusion in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: a randomized clinical trial. Stroke 46:3277–3281. https://doi.org/10.1161/strokeaha.115.010537
Ghanem MA, Shabana AM (2014) Effects of milrinone continuous intravenous infusion on global cerebral oxygenation and cerebral vasospasm after cerebral aneurysm surgical clipping. J Anaesth. https://doi.org/10.1016/j.egja.2013.07.006
Grubb S, Cai C, Hald BO, Khennouf L, Murmu RP, Jensen AGK, Fordsmann J, Zambach S, Lauritzen M (2020) Precapillary sphincters maintain perfusion in the cerebral cortex. Nat Commun 11:395. https://doi.org/10.1038/s41467-020-14330-z
Haegens NM, Gathier CS, Horn J, Coert BA, Verbaan D, van den Bergh WM (2018) Induced hypertension in preventing cerebral infarction in delayed cerebral ischemia after subarachnoid hemorrhage. Stroke 49:2630–2636. https://doi.org/10.1161/strokeaha.118.022310
Harik SI, Sharma VK, Wetherbee JR, Warren RH, Banerjee SP (1981) Adrenergic and cholinergic receptors of cerebral microvessels. J Cereb Blood Flow Metab 1:329–338. https://doi.org/10.1038/jcbfm.1981.36
Hayashida N, Tomoeda H, Oda T, Tayama E, Chihara S, Kawara T, Aoyagi S (1999) Inhibitory effect of milrinone on cytokine production after cardiopulmonary bypass. Ann Thorac Surg 68:1661–1667. https://doi.org/10.1016/S0003-4975(99)00716-X
Higgins JPT, Cochrane Collaboration (2020) Cochrane handbook for systematic reviews of interventions, Second edn. Wiley-Blackwell, Hoboken, NJ
Honerjäger P (1991) Pharmacology of bipyridine phosphodiesterase III inhibitors. Am Heart J 121:1939–1944. https://doi.org/10.1016/0002-8703(91)90828-6
Iida H, Iida M, Takenaka M, Oda A, Uchida M, Fujiwara H, Dohi S (2001) The effects of alpha-human atrial natriuretic peptide and milrinone on pial vessels during blood-brain barrier disruption in rabbits. Anesth Analg 93:177–182. https://doi.org/10.1097/00000539-200107000-00035
Jabbarli R, Pierscianek D, Rölz R, Oppong MD, Kaier K, Shah M, Taschner C, Mönninghoff C, Urbach H, Beck J, Sure U, Forsting M (2019) Endovascular treatment of cerebral vasospasm after subarachnoid hemorrhage: more is more. Neurology 93:e458–e466. https://doi.org/10.1212/wnl.0000000000007862
Jaeger M, Soehle M, Schuhmann MU, Meixensberger J (2012) Clinical significance of impaired cerebrovascular autoregulation after severe aneurysmal subarachnoid hemorrhage. Stroke 43:2097–2101
Jeng JS, Yip PK, Huang SJ, Kao MC (1999) Changes in hemodynamics of the carotid and middle cerebral arteries before and after endoscopic sympathectomy in patients with palmar hyperhidrosis: preliminary results. J Neurosurg 90:463–467. https://doi.org/10.3171/jns.1999.90.3.0463
Jespersen SN, Østergaard L (2012) The roles of cerebral blood flow, capillary transit time heterogeneity, and oxygen tension in brain oxygenation and metabolism. J Cereb Blood Flow Metab 32:264–277. https://doi.org/10.1038/jcbfm.2011.153
Joannides R, Haefeli WE, Linder L, Richard V, Bakkali EH, Thuillez C, Luscher TF (1995) Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation 91:1314–1319. https://doi.org/10.1161/01.cir.91.5.1314
Kajita Y, Dietrich HH, Dacey RGJ (1996) Effects of oxyhemoglobin on local and propagated vasodilatory responses induced by adenosine, adenosine diphosphate, and adenosine triphosphate in rat cerebral arterioles. J Neurosurg 85:908–916. https://doi.org/10.3171/jns.1996.85.5.0908
Kauffman RF, Schenck KW, Utterback BG, Crowe VG, Cohen ML (1987) In vitro vascular relaxation by new inotropic agents: relationship to phosphodiesterase inhibition and cyclic nucleotides. J Pharmacol Exp Ther 242:864–872
Khennouf L, Gesslein B, Brazhe A, Octeau JC, Kutuzov N, Khakh BS, Lauritzen M (2018) Active role of capillary pericytes during stimulation-induced activity and spreading depolarization. Brain 141:2032–2046. https://doi.org/10.1093/brain/awy143
Kim DH, Joseph M, Ziadi S, Nates J, Dannenbaum M, Malkoff M (2003) Increases in cardiac output can reverse flow deficits from vasospasm independent of blood pressure: a study using xenon computed tomographic measurement of cerebral blood flow. Clin Neurosurg 53:1044–1052. https://doi.org/10.1227/01.neu.0000088567.59324.78
Komas N, Lugnier C, Stoclet JC (1991) Endothelium-dependent and independent relaxation of the rat aorta by cyclic nucleotide phosphodiesterase inhibitors. Br J Pharmacol 104:495–503. https://doi.org/10.1111/j.1476-5381.1991.tb12457.x
Lannes M, Teitelbaum J, Cortés M d P, Cardoso M, Angle M (2012) Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital Protocol. Neurocrit Care 16:354–362. https://doi.org/10.1007/s12028-012-9701-5
Laskowitz DT, Kolls BJ (2010) Neuroprotection in Subarachnoid Hemorrhage. Stroke 41:S79–S84. https://doi.org/10.1161/STROKEAHA.110.595090
Levy ML, Rabb CH, Zelman V, Giannotta SL (1993) Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 79:494–499. https://doi.org/10.3171/jns.1993.79.4.0494
Macdonald RL, Pluta RM, Zhang JH (2007) Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Rev Neurol 3:256–263. https://doi.org/10.1038/ncpneuro0490
Macdonald RL, Weir B, Zhang J, Marton LS, Sajdak M, Johns LM (1997) Adenosine triphosphate and hemoglobin in vasospastic monkeys. Neurosurg Focus 3:e3
Miller JA, Jr RGD, Diringer MN (1995) Safety of hypertensive hypervolemic therapy with phenylephrine in the treatment of delayed ischemic deficits after subarachnoid hemorrhage. Stroke 26:2260–2266. https://doi.org/10.1161/01.str.26.12.2260
Moher D, Liberati A, Tetzlaff J, Altman DG (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ 339. https://doi.org/10.1136/bmj.b2535
Molnar-Kimber K, Yonno L, Heaslip R, Weichman B (1993) Modulation of TNFα and IL-1β from endotoxin-stimulated monocytes by selective PDE isozyme inhibitors. Agents Actions 39:C77–C79. https://doi.org/10.1007/BF01972726
Murphy A, Manoel AL d O, Macdonald RL, Baker A, Lee T-Y, Marotta T, Montanera W, Aviv R, Bharatha A (2017) Changes in cerebral perfusion with induced hypertension in aneurysmal subarachnoid hemorrhage: a pilot and feasibility study. Neurocrit Care 27:3–10. https://doi.org/10.1007/s12028-017-0379-6
Murray KJ (1990) Cyclic AMP and mechanisms of vasolidation. Pharmacol Ther 47:329–345. https://doi.org/10.1016/0163-7258(90)90060-F
Mutoh T, Ishikawa T, Kazumata K, Matsumoto K, Taki Y, Suzuki A (2014) Dobutamine versus mirlinone for intensive hemodynamic augmentation to relieve clinical delayed cerebral ischemia after subarachnoid hemorrhage. Stroke
Mutoh T, Mutoh T, Nakamura K, Yamamoto Y, Tsuru Y, Tsubone H, Ishikawa T, Taki Y (2017) Acute cardiac support with intravenous milrinone promotes recovery from early brain injury in a murine model of severe subarachnoid haemorrhage. Clin Exp Pharmacol Physiol 44:463–469. https://doi.org/10.1111/1440-1681.12718
Mutoh T, Mutoh T, Sasaki K, Nakamura K, Tatewaki Y, Ishikawa T, Taki Y (2017) Neurocardiac protection with milrinone for restoring acute cerebral hypoperfusion and delayed ischemic injury after experimental subarachnoid hemorrhage. Neurosci Lett 640:70–75. https://doi.org/10.1016/j.neulet.2017.01.008
Nakai K, Itakura T, Naka Y, Nakakita K, Kamei I, Imai H, Yokote H, Komai N (1986) The distribution of adrenergic receptors in cerebral blood vessels: an autoradiographic study. Brain Res 381:148–152. https://doi.org/10.1016/0006-8993(86)90703-1
Nishiguchi M, Ono S, Iseda K, Manabe H, Hishikawa T, Date I (2010) Effect of vasodilation by milrinone, a phosphodiesterase III inhibitor, on vasospastic arteries after a subarachnoid hemorrhage in vitro and in vivo: effectiveness of cisternal injection of milrinone. Neurosurgery 66:158–164. https://doi.org/10.1227/01.neu.0000363153.62579.ff
Nolan CP, Macdonald RL (2006) Can angiographic vasospasm be used as a surrogate marker in evaluating therapeutic interventions for cerebral vasospasm? Neurosurg Focus 21:E1–E8. https://doi.org/10.3171/foc.2006.21.3.1
Ohman J, Servo A, Heiskanen O (1991) Risks factors for cerebral infarction in good-grade patients after aneurysmal subarachnoid hemorrhage and surgery: a prospective study. J Neurosurg 74:14–20. https://doi.org/10.3171/jns.1991.74.1.0014
de Oliveira Manoel AL, Goffi A, Marotta TR, Schweizer TA, Abrahamson S, Macdonald RL (2016) The critical care management of poor-grade subarachnoid haemorrhage. Crit Care 20:21. https://doi.org/10.1186/s13054-016-1193-9
Orstavik O, Ata SH, Riise J, Dahl CP, Andersen GO, Levy FO, Skomedal T, Osnes J-B, Qvigstad E (2014) Inhibition of phosphodiesterase-3 by levosimendan is sufficient to account for its inotropic effect in failing human heart. Br J Pharmacol 171:5169–5181. https://doi.org/10.1111/bph.12647
Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K (1990) Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir 103:18–26. https://doi.org/10.1007/bf01420187
Painter PR, Edén P, Bengtsson H-U (2006) Pulsatile blood flow, shear force, energy dissipation and Murray’s Law. Theor Biol Med Model 3:31–10. https://doi.org/10.1186/1742-4682-3-31
Pegoli M, Mandrekar J, Rabinstein AA, Lanzino G (2015) Predictors of excellent functional outcome in aneurysmal subarachnoid hemorrhage. J Neurosurg 122:414–418. https://doi.org/10.3171/2014.10.JNS14290
Pluta RM (2005) Delayed cerebral vasospasm and nitric oxide: review, new hypothesis, and proposed treatment. Pharmacol Ther 105:23–56. https://doi.org/10.1016/j.pharmthera.2004.10.002
Porter VA, Bonev AD, Knot HJ, Heppner TJ, Stevenson AS, Kleppisch T, Lederer WJ, Nelson MT (1998) Frequency modulation of Ca 2+ sparks is involved in regulation of arterial diameter by cyclic nucleotides. Am J Phys Cell Phys 274:C1346–C1355. https://doi.org/10.1152/ajpcell.1998.274.5.C1346
Qureshi AI, Ishfaq A, Ishfaq MF, Pandhi A, Ahmed SI, Singh S, Kerro A, Krishnan R, Deep A, Georgiadis AL (2018) Therapeutic benefit of cilostazol in patients with aneurysmal subarachnoid hemorrhage: a meta-analysis of randomized and nonrandomized studies. J Vasc 10:33–40
Qureshi AI, Suarez JI, Bhardwaj A, Yahia AM, Tamargo RJ, Ulatowski JA (2000) Early predictors of outcome in patients receiving hypervolemic and hypertensive therapy for symptomatic vasospasm after subarachnoid hemorrhage. Crit Care Med 28:824–829. https://doi.org/10.1097/00003246-200003000-00035
Raabe A, Beck J, Keller M, Vatter H, Zimmermann M, Seifert V (2005) Relative importance of hypertension compared with hypervolemia for increasing cerebral oxygenation in patients with cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg 103:974–981. https://doi.org/10.3171/jns.2005.103.6.0974
Rouanet C, Reges D, Rocha E, Gagliardi V, Silva G (2017) Treatment with milrinone of delayed cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage: a tertiary academic hospital experience. Eur Stroke J. https://doi.org/10.1177/2396987317705242
Roy B, McCullough LD, Dhar R, Grady J, Wang Y-B, Brown RJ (2017) Comparison of initial vasopressors used for delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Cerebrovasc Dis 43:266–271. https://doi.org/10.1159/000458536
Saklani R, Jaggi A, Singh N (2010) Pharmacological preconditioning by milrinone: memory preserving and neuroprotective effect in ischemia-reperfusion injury in mice. Arch Pharm Res 33:1049–1057. https://doi.org/10.1007/s12272-010-0711-6
Sanelli PC, Kishore S, Gupta A, Mangat H, Rosengart A, Kamel H, Segal A (2014) Delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage: proposal of an evidence-based combined clinical and imaging reference standard. AJNR Am J Neuroradiol 35:2209–2214. https://doi.org/10.3174/ajnr.A3782
Sato K, Yoshimoto A (2000) Effects of olprinone on systemic and cerebral circulation in patients with subarachnoid hemorrhage. J Neurosurg Anesthesiol 12:81–83
Schmidt JM, Wartenberg KE, Fernandez A, Claassen J, Rincon F, Ostapkovich ND, Badjatia N, Parra A, Connolly ES, Mayer SA (2008) Frequency and clinical impact of asymptomatic cerebral infarction due to vasospasm after subarachnoid hemorrhage. J Neurosurg 109:1052–1059. https://doi.org/10.3171/JNS.2008.109.12.1052
Shan T, Zhang T, Qian W, Ma L, Li H, You C, Xie X (2019) Effectiveness and feasibility of cilostazol in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. J Neurol. https://doi.org/10.1007/s00415-019-09198-z
Shenkin HA (1969) Cervical sympathectomy on patients with occlusive cerebrovascular disease. Arch Surg 98:317–320. https://doi.org/10.1001/archsurg.1969.01340090093015
Shenkin HA, Cabieses F, Van Den Noordt G (1951) The effect of bilateral stellectomy upon the cerebral circulation of man. J Clin Invest 30:90–93. https://doi.org/10.1172/JCI102421
Slim K, Nini E, Forestier D, Kwiatkowski F, Panis Y, Chipponi J Methodological index for non-randomized studies (MINORS): development and validation of a new instrument. 5. https://doi.org/10.1046/j.1445-2197.2003.02748.x
Soliman R, Zohry G (2019) Effect of magnesium sulphate and milrinone on cerebral vasospasm after aneurysmal subarachnoid hemorrhage: a randomized study. J Anesth. https://doi.org/10.1016/j.bjan.2018.09.005
Stein SC, Levine JM, Nagpal S, LeRoux PD (2006) Vasospasm as the sole cause of cerebral ischemia: how strong is the evidence? Neurosurg Focus 21:1–7. https://doi.org/10.3171/foc.2006.21.3.2
Sulek CA, Blas ML, Lobato EB (2002) Milrinone increases middle cerebral artery blood flow velocity after cardiopulmonary bypass. J Cardiothorac Vasc Anesth 16:64–69. https://doi.org/10.1053/jcan.2002.29680
Suzuki J, Iwabuchi T, Hori S (1975) Cervical sympathectomy for cerebral vasospasm after aneurysm rupture. Neurol Med Chir (Tokyo) 15(pt 1):41–50. https://doi.org/10.2176/nmc.15pt1.41
Tranmer BI, Keller TS, Kindt GW, Archer D (1992) Loss of cerebral regulation during cardiac output variations in focal cerebral ischemia. J Neurosurg 77:253–259. https://doi.org/10.3171/jns.1992.77.2.0253
Tsukahara T, Taniguchi T, Shimohama S, Fujiwara M, Handa H (1986) Characterization of beta adrenergic receptors in human cerebral arteries and alteration of the receptors after subarachnoid hemorrhage. Stroke 17:202–207. https://doi.org/10.1161/01.STR.17.2.202
Vas N, Beigh A, Alabdulraheem N, Bosnjakovic P, Abulhasan Y (2013) Targeted intra-arterial milrinone for the treatment of symptomatic cerebral vasospasm in aneurysmal sah. Neurocritical Care. https://doi.org/10.1007/s12028-013-9895-1
Vergouwen MDI, Vermeulen M, van Gijn J, Rinkel GJE, Wijdicks EF, Muizelaar JP, Mendelow AD, Juvela S, Yonas H, Terbrugge KG, Macdonald RL, Diringer MN, Broderick JP, Dreier JP, Roos YBWEM (2010) Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 41:2391–2395. https://doi.org/10.1161/STROKEAHA.110.589275
Washington CW, Derdeyn CP, Dhar R, Arias EJ, Chicoine MR, Cross DT, Dacey RGJ, Han BH, Moran CJ, Rich KM, Vellimana AK, Zipfel GJ (2016) A phase I proof-of-concept and safety trial of sildenafil to treat cerebral vasospasm following subarachnoid hemorrhage. J Neurosurg 124:318–327. https://doi.org/10.3171/2015.2.JNS142752
Young RA, Ward A (1988) Milrinone: a preliminary review of its pharmacological properties and therapeutic use. Drugs 36:158–192. https://doi.org/10.2165/00003495-198836020-00003
Yukiiri K, Mizushige K, Ueda T, Nishiyama Y, Aoyama T, Kohno M (2001) Effects of olprinone, a phosphodiesterase 3 inhibitor, on regional cerebral blood flow of cerebral cortex in stroke patients. J Cardiovasc Pharmacol 37:375–380. https://doi.org/10.1097/00005344-200104000-00004
Milrinone - Full product Information. In: MIMS Online. https://www.mimsonline.com.au.acs.hcn.com.au/Search/FullPI.aspx?ModuleName=Product%20Info&searchKeyword=milrinone&PreviousPage=~/Search/QuickSearch.aspx&SearchType=&ID=38220001_2. Accessed 6 Jun 2020
Author information
Authors and Affiliations
Contributions
MCK collected and analysed the data, wrote the draft, and revised the manuscript. LL helped analyse the data and revise the manuscript. JM, HA, RAD, and TG provided critical interpretation of the data and helped revise the manuscript. RVC helped analyse the data, revised the manuscript, and provided study supervision. All authors read and approved the final manuscript. All authors approved the final submission, and all authors agree to be accountable for all aspects of the work
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 18 kb)
Rights and permissions
About this article

Cite this article
Castle-Kirszbaum, M., Lai, L., Maingard, J. et al. Intravenous milrinone for treatment of delayed cerebral ischaemia following subarachnoid haemorrhage: a pooled systematic review. Neurosurg Rev 44, 3107–3124 (2021). https://doi.org/10.1007/s10143-021-01509-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10143-021-01509-1