
Abstract
Central neurocytomas (CN) are uncommon tumors of the central nervous system, most descriptions of which available in the literature are in the form of isolated case reports and small series. Owing to this rare incidence, diagnosis and management of this neoplasm remain controversial. Usually, these tumors affect lateral ventricles of young adults and display characteristic neuroimaging and histomorphologic findings. Neurocytomas often mimic oligodendrogliomas when confirmation of diagnosis rests on immunohistochemistry, ultrastructure, and genetic studies. Extraventricular neurocytomas, situated entirely within the brain parenchyma and spinal cord, have also been reported. Typically, CN are associated with a favorable outcome although cases with more aggressive clinical course with recurrences are not unknown. MIB-1 labeling index (LI) of >2% often heralds poor prognosis and tumour recurrence. Safe maximal resection is presently considered the ideal therapeutic option, with best long-term prognosis in terms of local control and survival. The role of adjuvant radiotherapy apparently seems to benefit patients with incomplete resection and in atypical neurocytoma. Utility of other therapeutic regimen, however, remains shrouded in controversy. Epidemiology, histogenesis, clinical profile, histology, neuroimaging and therapeutic modalities of neurocytomas have been comprehensively reviewed, with special emphasis on CN and extraventricular neurocytomas and their atypical counterparts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Central neurocytomas (CN) corresponding to World Health Organization (WHO) grade II [31] are uncommon, benign tumors of the central nervous system (CNS) associated with favorable outcome. Historically, many such lesions were regarded as intraventricular oligodendrogliomas or ependymomas until detailed immunocytochemical clarification of their neuronal phenotype was established. Currently, this entity has been well recognized outside the confines of the cerebral ventricular system [37] and have been termed as “extraventricular neurocytomas” (EVN). Thus, more important than classifying these rare neuronal tumors based on their location, it is imperative to recognize neurocytomas as a group and not mistake them for infiltrating gliomas, such as oligodendrogliomas, which they mimic due to the perinuclear halos acquired during the excision-fixation interval.
Histogenesis
Since the original description by Hassoun et al. [45], it has generally been accepted that CN develop from the nuclei of the septum pellucidum. Subsequently, Nishio et al. [83], von Deimling et al. [129] and other workers [51] have extrapolated this hypothesis to include the third ventricle and the subependymal plate of the lateral ventricles.
Currently, it remains controversial as to whether neurocytomas arise from cells committed to neuronal phenotype [30] or from bipotential cells in the periventricular matrix [126]. In vitro cultures by Westphal et al. [131] and several others [6, 51, 52] have revealed that CN have properties reminiscent of bipotential precursor cells, which can exhibit both glial and neuronal differentiation. A recent report by You et al. [136] further proposed that CN cells can differentiate into neuronal cells in vivo and into glial cells in vitro. This bipotential property possibly explains the glial fibrillary acidic protein (GFAP)-immunopositive cell population observed in true CN [51, 101, 112, 128, 129, 132] and may also account for detection of spinal CN arising from the central canal region [113, 117].
Incidence, age, and gender distribution
Although CN has gained considerable recognition following the first report by Hassoun et al. [45] in 1982, this continues to remain an uncommon entity, comprising only 0.1–0.5% of all brain neoplasms [18, 31, 46, 60], with most available literature being in the form of isolated case reports and small series. Apparently, there appears to be a genetic difference between Asians and Caucasians, which is apparent from the frequent reporting of these cases from Korea [57–61], India [63, 107–109], and Japan [51, 52, 56, 62, 79, 80, 85, 124].
Typically, CN affects young adults around the third decade [31, 46], with cases ranging from 8 days to 67 years of age [31]. Apart from Sharma et al. [108] and Kim et al. [57] who have noted male predominance in their series, most studies have observed equal affection of both genders [46]. Extraventricular neurocytomas, first reported in 1989 [29], are more unusual tumors. They have generally been observed to afflict children and young adults, the age range being between 5 and 76 years (median 34 years), with no gender predilection.
Location
Most neurocytomas are midline supratentorial lesions straddling the lateral and third ventricles, often in relation to the foramen of Monro and arising from the septum pellucidum, fornix, or walls of the lateral ventricles [45, 46]. In their review of 127 cases, Hassoun et al. [46] noted involvement of the lateral ventricle and third ventricle in 98 and 33 cases, respectively, with 27 of the latter group showing lateral ventricular extension. Interestingly, right-sided lesions were more common, which was contrary to that observed by other series [31]. EVN, on the contrary, are primary tumors of the brain parenchyma devoid of any apparent connection with the ventricular system. Case reports have documented involvement of cerebral hemispheres (commonly frontal followed by parietal) [11], thalamus [106], cerebellum [88], pons [111], amygdala [92], pineal gland [82], retina [74], and spinal cord [18, 71, 109, 113, 117]. Interestingly, presence of neurocytoma has sometimes been observed arising outside the CNS, such as an ovarian teratoma [48], and in the pelvic cavity [32].
Presenting features
CN patients have a slow and benign clinical course and manifest with features of raised intracranial pressure secondary to obstructive hydrocephalus caused by impaired cerebrospinal fluid (CSF) flow or with seizures, headache, nausea, vomiting, and memory and visual disturbances [1, 11, 46] as a result of the mass effect of the tumor [46, 76]. Cases with more acute presentation due to hemorrhage into the tumor are also well described [110, 117]. EVN generally manifest through mass effects in the form of seizures and hemiparesis [125]. Brat et al. [11] described a series of 35 cases of cerebral EVN that presented with seizures, diplopia, headache and vomiting. Neurocytomas of the spinal cord present with paraesthesia, numbness, and weakness of limbs [71, 109, 113, 117] depending on the level of affection.
Neuroimaging
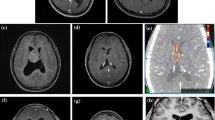
Imaging (Fig. 1) generally shows CN as discrete, solitary, masses near the foramen of Monro, most of which are attached to the septum pellucidum and expand into one or both lateral ventricles and, less commonly, into the third ventricle. Sequential scans demonstrate slow growth. On computed tomography (CT) scan, these are either isodense or mildly hyperdense, with strong, generally uniform, contrast enhancement. Calcifications and cystic changes have also been reported. Magnetic resonance (MR) imaging shows high signal on both T1- and T2-weighted images, with moderate to strong enhancement after gadolinium administration [31, 108].

a Axial contrast-enhanced computed tomography (CT) scan shows an intraventricular mass lesion in the body of the right lateral ventricle with heterogenous density. b T1-weighted contrast-enhanced coronal magnetic resonance (MR) image shows a heterogenous enhancing mass lesion in the left ventricle
Radiologically, EVN (Fig. 2) are generally discrete, sometimes large, complex and variably enhancing masses. These are often cystic, frequently calcified, with or without associated hemorrhage [5–7]. Spinal neurocytomas are generally visualized as intramedullary solitary lesions, isointense on T1- and hyperintense on T2-weighted images [11, 109]. Occasionally, these may mimic spinal astrocytomas (Fig. 2d).

a Axial computed tomography (CT) scan shows juxtaventricular mass lesion in the right frontal lobe with cystic and solid component, along with calcification. b T1-weighted contrast-enhanced magnetic resonance (MR) image showing dense enhancement in the solid component. Note that the cyst wall does not show any enhancement, and there is minimal perilesional edema. c Postgadolinium sagittal T1-weighted image shows densely enhancing intramedullary lesion at C5–C6 level. d Sagittal T2-weighted image showing intramedullary mass lesion extending from C7 to D8, with cord expansion and hyperintense signal mimicking astrocytomas
Cerebral angiography is rarely performed, which shows homogenous vascular blush persisting till late venous phase [31, 36, 112]. Jayasundar et al. [55] performed in vivo and in vitro proton MR spectroscopic (MRS) study on two cases and suggested presence of N-acetyl aspartate (NAA) with increased glycine as possible characteristic feature of CN. However, a recent study by Chuang et al. [16] observed increased choline/creatine ratios and decreased NAA/creatine ratios in all three cases, with alanine peak signifying poor prognosis. Glycine peak may possibly suggest CN, but its absence does not exclude the diagnosis [55, 61]. Ando et al. [4] noted markedly elevated choline and lactate peaks with a strongly diminished N-acetyl aspartate peak.
Pathology
Macroscopic features
Neurocytomas are generally grayish in color, resembling the grey matter, with areas of hemorrhage. Grittiness experienced while dissecting resected specimen is due to presence of calcification [13, 31, 50].
Light microscopy
Central neurocytoma
CN are well-differentiated tumors with benign histology, which may display varied histological architecture even in the same specimen. The honeycomb arrangement of oligodendroglioma, and large fibrillary areas resembling irregular rosettes of pineocytoma are the commonest patterns identified in CN. In other areas, cells may be arranged in straight lines or as perivascular rosettes mimicking ependymoma. These tumors are composed of uniform, small-to-medium-sized cells with rounded nuclei, finely stippled chromatin (“salt and pepper” chromatin) and inconspicuous nucleoli, along with scant cytoplasm (Fig. 3a,b). Cells are usually closely apposed but may also be set within a background of finely filamentous stroma having a neuropil-like quality. Hematoxylin and eosin (H&E) stained sections often reveal fixation artefacts in the form of cytoplasmic vacuolations, closely resembling the “fried-egg” appearance of oligodendroglioma (Fig. 3e). Vascularity is represented by long, thin-walled capillary-sized vessels, which are arranged in a linear arborizing pattern, imparting an endocrine appearance. In many cases, thin-walled dilated vascular channels (Fig. 3d), and foci of calcification (Fig. 3e) are readily identified. In places, tumors display dense cellular areas alternating with fibrillary/acellular areas. The latter components are mainly perivascular and have a fine fibrillary neuropil matrix mimicking “rosettes of ependymoma” (Fig. 3g,h). Calcifications in the form of small calcospherites distributed throughout the tumor parenchyma have been noted in half the biopsies [31] while rare cases exhibit features such as Homer-Wright rosettes, neuroblastic component, or ganglioid cells [99, 128].

Histomorphology of central neurocytoma (CN) showing sheets of uniform, small-to-medium-sized cells with rounded nuclei, finely stippled chromatin, and inconspicuous nucleoli, along with scant cytoplasm [a: hematoxylin and eosin (H&E)×100; b: H&E×200]. In places, the tumor displayed cellular areas alternating with fibrillary/acellular areas (c: H&E×100). Focal areas of calcification (d: H&E×100), along with thin-walled, dilated, vascular channels (e: H&E×40) were present in many cases. Interspersed between uniform cells with rounded nuclei were delicate, capillary-sized vascular channels forming branching network pattern, resembling oligodendroglioma (f: H&E×100). Certain cases, however, had to be differentiated from dysembryoplastic neuroepithelial tumors (DNT) (g: H&E×200). In some cases, cells had a perivascular arrangement and with fine fibrillary neuropil matrix mimicking “rosettes of ependymoma” (h: H&E×100). All cases were synaptophysin immunopositive (i: Syn×200) and showed glial fibrillary acidic protein (GFAP)-positive reactive astrocytes in the perivascular location (j: GFAP×200). MIB-1 labeling index (LI) was, however, low (k: MIB-1×100). Presence of neurosecretory granules on ultrastructural examination confirmed the neuronal origin of the tumors [l: electron microscopy (EM)×1,650]
Extraventricular neurocytoma
In contrast to CN (Table 1), typical EVN display a wider morphological spectrum encompassing arrangement of neurocytes in sheets, clusters, ribbons, or rosettes, with neuropil dispersed either in broad zones (neuropil islands) between these cell arrangements or localized within the rosettes [11]. Neurocytes most often demonstrate finely granular, slightly eosinophilic cytoplasm, occasionally with artefactual vacuolations resembling oligodendroglioma. Nuclei of these tumors are strikingly monotonous and rounded, with finely speckled chromatin, distinct nuclear rims, and one to three small nucleoli.
Ganglionic differentiation is more common in EVN [30, 31, 112, 128] and has been observed in 66% cases in one series [11]. These were either focal (29%) or diffuse (37%), with few cases showing compact nodules of ganglion cells against a neurocytic background [11]. Mitotic activity is uncommon while calcification is frequent. Cerebral EVN generally have pushing borders along with well-defined interface with adjacent brain parenchyma [11].
Other variants
After the initial description of neurocytoma, several variants have been described. Kim and Suh reported a case of pseudopapillary neurocytoma of the temporal lobe with glial differentiation [59] while others noted CN with predominance of ganglioid differentiation (ganglioneurocytoma) [35, 84]; with pigmentation [82]; or with a more pleomorphous component, viz. neurolipocytoma [2, 25, 49, 73], myoneurocytoma [88], and with both glial and neuronal differentiation [103]. In few cases, cortical neurocytomas presenting with seizures in young patients would otherwise correspond with complex dysembryoplastic neuroepithelial tumors (DNT) [20] or nonspecific DNT of Daumas-Duport et al. [21].
Immunohistochemistry
The vast majority of neurocytomas and neurocytoma-like lesions have been diagnosed on the basis of synaptophysin immunoreactivity (Fig. 3i) alone, even without ultrastructural evaluation [126]. Strong immunostaining for synaptophysin has been recognized as the most suitable and reliable diagnostic marker. Typically, synaptophysin immunoreactivity is noted in the neuropil, especially in fibrillary zones and perivascular cell-free areas [31], and not in the cell bodies of normal neurons [75]. False cytoplasmic immunopositivity may be attributed to preexistent neuropil or neuronal structures, or faulty antigen retrieval techniques and/or use of polyclonal antisynaptophysin antibody. On the contrary, false immunonegativity ascribed to spontaneous absence of immunoreactivity [47], small biopsies, or technical difficulties, does not necessarily rule out a diagnosis of neurocytoma [91]. However, in the absence of other supportive evidence, intracytoplasmic and paranuclear immunolabeling in EVN must be interpreted cautiously in order to avoid misinterpretation of false positive staining in oligodendroglioma.
Tumor cells have been reported to express an array of neuronal markers, including neuron-specific enolase (NSE), neuron-associated class III beta-tubulin, tau, microtubule-associated protein-2 (MAP2), calcineurin, protein gene product 9.5 (PGP 9.5) [30, 39, 46, 47, 113, 129], HNK1 (Leu7) [99], alpha-synuclein [56], neuron-specific antigen L1, and embryonal form of neural cell adhesion molecule (E-NCAM) [30]. Neuronal nuclear antigen (NeuN) expression is generally associated with tumor cells displaying terminal neuronal differentiation [44] and is often helpful in resolving ambiguous synaptophysin staining [11]. Interestingly, Hu-immunoreactivity is restricted to the nuclei of neurocytes only [40, 44], especially in those surrounding neuropil islands [44]. Chromogranin-A and neurofilament are usually absent, except when admixed with ganglion cells [46].
Expression of GFAP (Fig. 3j) has been observed more frequently in EVN compared with CN (Table 1) [11, 31]. It may be difficult to distinguish GFAP-immunopositive reactive [47] and entrapped nonneoplastic astrocytes from tumor cells showing focal cytoplasmic GFAP immunostaining with neurocytic features [11, 121, 136]. Though this bears testimony to origin from bipotential cells of germinal matrix, this distinction appears to bear little or no prognostic connotation.
An unusual case of cerebellar neurocytoma with immunohistochemically confirmed rhabdomyomatous differentiation has also been described [88].
Electron microscopy
Electron microscopy is imperative in a few cases, either to resolve diagnostic dilemmas evolving from doubtful synaptophysin immunostaining or to demonstrate unequivocal neuronal features in EVN [11, 31, 51, 62]. Ultrastructural evaluation (Fig. 3l) establishes neuronal origin of these tumors, including cell processes containing parallel arrays of microtubules, with both clear vesicles and dense-core granules in their terminations. Though intercellular junctions and rare synapses may be seen, these are not essential for diagnosis [11, 31, 45, 51, 62, 99].
Proliferation and grading
Most neurocytomas are cytologically bland and mitotically deprived and correspond to WHO grade II [31]. Occasional cases, however, display histologic features of anaplasia or malignancy without clinical evidence of poor outcome [68, 107], for which Mackenzie et al. [68] reserved the designation “proliferating neurocytoma.” Typically, proliferative index of neurocytomas as evaluated by silver colloidal staining for argyrophilic nucleolar organizer regions (AgNOR counts) or by Ki67 and proliferating cell nuclear antigen (PCNA) immunolabeling have been low [42, 57]. Currently, MIB-1 labeling index (MIB-1 LI) is routinely used to assess proliferation potential of neurocytomas. Kim et al. [57] revealed diploidy in all their cases of neurocytomas using DNA flow cytometry. A recent double labeling immunohistochemistry study by Englund et al. [27] observed that NeuN expression correlates with reduced mitotic index (as measured by Ki-67) of neoplastic cells in CN.
Atypical neurocytoma
The terms “atypical neurocytoma”/“atypical extraventricular neurocytoma” have been coined for neurocytoma/EVN exhibiting MIB-1 LI >2% [26, 33, 112, 123, 132] with/without anaplastic features viz. focal necrosis, vascular proliferation, and increased mitotic activity (Fig. 4). Cases with MIB-1 LI >2% (Fig. 4d) have been noted to predict adverse outcome [68, 112] in terms of time to recurrence or progression after surgery. However, a recent evaluation by Rades et al. [97] has suggested a MIB-1 index score >3% is associated with poor clinical outcome.

a–d Histopathology of atypical spinal cord neurocytoma [a: hematoxylin and eosin (H&E)×200 showing mitosis (inset A →: H&E×400)] along with areas of necrosis (b: H&E×100) and endothelial proliferation (c: H&E×100). MIB1 labeling index (LI) was raised (d: MIB-1×200)
Molecular genetics
Cytogenetic and molecular studies for neurocytomas have yielded variable and inconsistent results in the form of loss of chromosome 17 [15]; gain of chromosomes 2p, 10q, 18q [135], and 7 [116]; and isochromosome 17 [16, 54]. However, TP53 and v-myc myelocytomatosis viral related oncogene, neuroblastoma derived (avian) (MYCN) amplification [54, 86, 129] and loss of heterozygosity of chromosome 1p/19q were consistently not detectable in these tumors, the latter feature proving diagnostic in differentiating from oligodendrogliomas [34, 119].
Differential diagnosis
Oligodendroglioma
Owing to their artefactual cytoplasmic vacuolations and monomorphic histologic appearances, neurocytomas, especially EVN, need to be differentiated from oligodendroglial tumors [13, 31, 50, 78]. Awareness of the condition, an intraventricular localization, along with histologic features of cellular uniformity, neuropil islands, streaming of cells, salt and pepper chromatin, ganglionic differentiation (more in EVN), eosinophilic granular bodies, and lack of entrapped or infiltrated brain parenchyma, suggest a diagnosis of neurocytoma [11, 31]. Immunoreactivity for synaptophysin in neurocytomas substantiates histology. Subsequent confirmation can be achieved by observing dense core granules and microtubules on electron microscopy, lack of p53 immunostaining, and absence of chromosome 1p/19q loss (Table 2).
Ependymoma
An intraventricular tumor with solid, noninfiltrating pattern along with perivascular pseudorosettes warrants a differential diagnosis of ependymomas. However, characteristic features of nuclear roundness with speckled chromatin and comparatively ill-defined perivascular rosettes suggest neurocytic origin, which are confirmed by synaptophysin immunopositivity of rosettes along with GFAP immunonegativity [13, 31, 50].
Dysembryoplastic neuroepithelial tumor (DNT) (Fig. 2f)
Both DNT and neurocytomas are neuronal/glioneuronal tumors characterized by uniform nuclei and perinuclear halo, with the former being less cellular and more mucoid on histology [20–22].
Others
Conditions such as cerebellar “medullocytomas” or “lipidized medulloblastomas” are differentiated from more ominous medulloblastomas by virtue of their occurrence in adults, bland cytomorphology, and impoverished mitotic activity [89]. Cerebellar liponeurocytoma is another rare and recently identified tumor characterized by areas of lipomatous differentiation, with apparently a favorable prognosis. Histologically, these show focal accumulation of adipose tissue in an otherwise typical small cell tumor and resemble CN [2, 73].
Differential diagnosis of spinal neurocytomas
Differential diagnoses of spinal neurocytomas (SN) include ependymomas, astrocytomas, oligodendrogliomas, mixed gliomas, neuroblastomas, and hemangioblastomas, and, rarely, metastasis [3, 5]. Unlike SN, neuroblastomas are small round-cell tumors composed of densely packed anaplastic cells, with hyperchromatic nuclei, very scant cytoplasm, frequent mitosis and an intraparenchymal location.
Therapeutic strategies
Surgical therapy
Most neurocytomas are intraventricular in location without any invasion into the surrounding brain parenchyma, thus being amenable to microsurgical macroscopic total resection. Surgery not only provides tissue for histological diagnosis but also helps reestablish CSF pathways and enable subsequent safe resection [102]. Various surgical approaches have been adopted for resection of CN, the most common being transcortical-transventricular and interhemispheric-transcallosal-transventricular routes. Since most CN straddle the lateral and third ventricle, a transcallosal approach provides the greatest flexibility. The transcortical route finds favor with one-sided tumors while the contralateral interhemispheric approach appears more conducive for neoplasms occurring superiorly on one side of the ventricular system [102]. Despite multiple interventions, resection may remain incomplete due to adhesions to eloquent structures such as the fornix. These are caused by profuse intraoperative bleeding and excessive calcifications [9], thus rendering gross total resection (GTR) difficult. In the compilation of 310 cases reported by Rades et al. [93, 95], GTR was possible in 44% while Bertalanffy et al. [9] achieved GTR in 57% among their series of 14 patients. Surgery helps to reestablish CSF pathways and alleviates raised intracranial pressure. External ventricular drainage as a temporary measure prior to definite microsurgery is reserved for patients with rapidly declining neurologic status and evidence of hydrocephalus. A permanent shunt, however, is considered for patients who continue to have hydrocephalus while a third ventriculostomy is considered for cases with noncommunicating hydrocephalus [14, 102]. Buxton et al. [14] noted successful endoscopic third ventriculostomy in 80% of their 63 patients with noncommunicating hydrocephalus while that for the intraventricular tumor group was 86%.
Surgical experiences with spinal neurocytomas have been variable. Both extramedullary and intramedullary tumors were amenable to complete removal while a highly vascular SN could not be removed at first attempt owing to focal infiltrative component [5, 18, 114]. Subsequent radiotherapy (RT) followed by complete resection precipitated significant neurological deficit [117]. However, precautions need to be taken during GTR to avoid vascular compromise to the remaining spinal cord [117].
Radiotherapy
Owing to the small but demonstrable risk associated with surgery, alternative/adjuvant modalities in the form of RT have been pursued, especially in a setting of subtotal resection (STR) and/or recurrence [3, 8, 10, 17, 23, 57, 58, 60, 79, 101, 125, 130, 134]. However, it is unanimously agreed that considering the benign nature of the tumor and risks of radiation toxicity, routine prophylactic radiotherapy be avoided in those cases with complete resection only and in cases that are not atypical. [93, 95].
Owing to the paucity of cases and lack of adequate follow-up, role of RT as primary treatment modality after biopsy remains controversial. However, while managing small tumors in setups lacking stereotactic surgery, this may be an option, provided a regular follow-up for recurrence is possible. Kulkarni et al. [63] studied the role of whole-brain irradiation in seven patients after stereotactic biopsy. None of the cases experienced any procedure-related adverse effects, but one had disseminated intracranial disease at 15 months while the other six were symptom-free with local control at 78 months of follow-up. In one case, Kim et al. [58] also noted demyelinating changes in the brain parenchyma adjacent to the tumour covered by the field of radiation therapy. Further, Utsunomiya et al. [124] reported recurrence in the form of anaplastic astrocytoma eight years after initial treatment by partial resection and RT in a case of central neurocytoma.
Role of RT as adjuvant following GTR remains debatable since this subset of patients already have a better local control. Rades and Fehlauer [93, 95] noted no additional benefit of RT following GTR in terms of improved local control and increased survival for both typical and atypical CN. In contrast, RT after STR in typical CN showed improved local control but not survival while in atypical CN, adjuvant RT made a marked difference in improving both parameters.
In a retrospective analysis of patients receiving RT, Rades et al. [98] noted 54 Gy or greater (in 2-Gy fraction) being the optimal radiation dose in terms of providing highest chance of local control and longest time to recurrence. It has been agreed upon that adverse effects due to radiation toxicity are minimal if the total radiation does not exceed 60 Gy [93, 95]. Currently, the optimal dose for RT for patients with CN seems to be 54—60 Gy. In most cases, tumor bed and adjacent parenchyma should be included in the RT field in order to achieve maximal effect [12, 93, 101].
Stereotactic radiosurgery
With the introduction of stereotactic radiosurgery, viz., linear accelerator (LINAC) and gamma knife surgery (GKS), it is currently possible to deliver high-dose radiation with minimal long-term side effects [67]. Initial experience with CN patients showed equivocal but encouraging results [3, 90], with hopes of providing options of a safe and effective adjuvant therapy following initial resection with or without recurrence and eliminating the need of a reoperation.
Gamma knife surgery (GKS)
Focused radiation enables precise delivery of high-dose in one singe-treatment session. In intraventricular tumors, excess radiation is borne by the surrounding CSF layer, thus reducing possible ill effects to the neighboring brain parenchyma [8]. Experience with GKS as a primary therapeutic option for CN is restricted to only two cases reported by Tyler-Kabara et al. [122] and a single case by Javendan et al. [53]. Though reduction in tumor size following GKS has been minimal, none of the cases experienced any side effects, thus necessitating the need for evaluating this as a primary treatment option after biopsy. Cobery et al. [17] used GKS as an adjuvant therapy for residual tumor after STR, as an alternative to conventional radiotherapy, in three cases while Hara et al. [43] introduced it in a single case. All four cases showed marked decrease in tumor size during a follow-up period ranging from 12 months to 99 months with minimal side effects. Many workers [3, 8, 17, 90, 101, 122] have used GKS as the primary therapeutic option for managing CN with asymptomatic recurrence. Overall, 15 out of 16 cases showed favorable response in the form of reduction in tumor volume during a follow-up period of 12–53 months. Most patients tolerated the procedure without any side effects and were discharged the next day, thus considerably reducing morbidity. In a recent study of recurrent CN, Bertalanffy et al. [9] noted a good response to different forms of focal radiation therapy (gamma knife radiosurgery in three patients and interstitial irradiation in one patient).
Linear accelerator (LINAC)
Although limited to only three reports, response to LINAC radiosurgery [51, 58, 70, 72] has been favorable. Kim et al. [58] utilized LINAC radiosurgery in the management of residual lesion. Tumor started regressing within 6 months and completely disappeared by 36 months, with no radiological sign of recurrence even after 51 months of the procedure. Martin et al. [70] utilized LINAC radiosurgery in the management of four patients with residual lesion after initial surgery. Two were cured, one showed considerable reduction in tumor size, and the fourth remained stable. They concluded that in cases of small residual tumors or recurrences, radiosurgery allows open surgery to be avoided and is a safe and potentially effective approach.
Cyberknife
Recently, a frameless image-guided stereotactic radiosurgery with Cyberknife (Accurav, Sunnyvale, CA, USA) has been developed, which, although not used in CN to date, may serve as a viable alternative to the available treatment regimen [65].
Chemotherapy
Chemotherapy has been found beneficial in recurrent CN that cannot be resected but have been irradiated without success [102]. It has usually constituted part of a multimodality therapeutic regimen as an acceptable alternative to potential long-term side effects of radiotherapy in younger patients. A wide variety of chemotherapeutic agents, including carmustine, lomustine, prednisone, vincristine, and cisplatin, have been used [10, 23, 130, 134]. Schild et al. [101] reported a series of four cases that received chemotherapy after radiation without any tumor progression. Dodds et al. [23] treated a 15 year old with chemotherapy comprising etoposide, ifosfamide, and carboplatin after initial STR. Though the tumor responded well to this regimen, the patient required reoperation and RT. They also evaluated the role of chemotherapy (etoposide, cisplatin, and cyclophosphamide) in recurrent CN patients who were initially managed with surgery with or without RT. Stabilization of disease was noted in two cases and complete remission in one [23]. Recently, von Koch and colleagues [130] described a case of a 20-year-old woman who received procarbazine, lomustine, and vincristine (PCV) chemotherapy after four STRs. Her tumor size started reducing after two cycles and continued to shrink until it stabilized after five cycles. Despite the limited but promising experience with chemotherapy in CN, larger cohorts of patients need to be evaluated prior to incorporating this in the conventional treatment schedule.
Biological behavior and outcome
CN are thought to represent the benign end of the spectrum of neuronal tumors that are generally amenable to surgical excision, with an overall favorable outcome. Even residual tumors following STR are noted to behave in a biologically indolent fashion. Eng et al. evaluated 145 cases of CN reported in the literature between 1982 and 1997 [26] and observed only seven recurrences, all of which were local [57, 60, 99, 106, 134]. In another series of 11 surgically resected patients, without adjuvant RT/chemotherapy, death could be attributed to tumor in only one case [37]. Schild et al. [101] evaluated a series of 32 CN, and noted that patients treated with GTR without adjuvant RT had a 5-year local control and survival rate of 100% and 80%, respectively. However, in STR cases, RT increased the control rate from 50% to 100%. Brown and colleagues [12] also found that crude local control rates for STR versus STR/RT (48.6–626.2 Gy in 180–200 cGy fractions or 59 Gy median dose) were 62% and 100% (p=0.0008), respectively.
Rades et al. [95] evaluated data of 310 CN patients from 91 different institutes and concluded that patients managed by GTR had a better outcome compared with STR, and adding RT to GTR did not improve local control or 5-year survival rate while STR-RT had better local control but not survival rate. In this study [95], they observed local control rates of 95% and 85% for GTR at 3 years and 5 years compared with 55% and 46% for STR. On introducing adjuvant RT, 3-year and 5-year local control rates for GTR remained at 96% and 86%, respectively, while it improved to 89% and 83%, respectively, with STR. The 5-year survival rates of 99% for GTR and 86% for STR did not show any improvement on adding RT and were comparable with the 95% for GTR-RT and 90% for STR-RT (Table 3).
Further, on evaluating 85 patients with atypical CN, Rades and colleagues [94] noted the significant impact of RT following STR whereby local control and survival rates improved significantly. Local control rates at 3 years and 5 years in this study were 73% and 57%, respectively, for GTR and 21% and 7%, respectively, for STR, with 3-year and 5-year survival rates being 93% and 93%, respectively, following GTR and 65% and 43%, respectively, after STR. On adding adjuvant RT, the 3-year and 5-year local control rates for GTR patients remained at 81% and 53%, respectively, while in STR cases, it improved to 85% and 70%, respectively. Similarly, survival at 3 years and 5 years also remained unchanged for GTR at 90% and 90%, respectively, while it enhanced for STR cases to 87% and 78%, respectively (Table 3).
Neurocytoma is an extremely rare tumor in the pediatric age group, with approximately 60 cases been documented since initial description [96]. A comprehensive review by Rades et al. [96] also showed better outcome with GTR compared with STR. Local control rates at 5 years and 10 years were 86% and 86%, respectively, for GTR in comparison with 60% and 45%, respectively, for STR. The 5- and 10-year survival rates, however, were 100% and 93%, respectively, after GTR and after STR. Despite encouraging results, RT has failed to find favor with many workers [58], and some have even cautioned against routine use of adjunctive RT following STR [26, 41, 62, 68, 69, 80, 83, 87, 105, 117, 127, 128] owing to the hazards of radiation toxicity, especially in the pediatric age group with a still-growing skeleton [113]. Some have, however, advocated use of RT for managing residual tumor based on MIB-1 LI [65, 94, 95, 101].
Despite the initial concept of benign behavior of CN, long-term follow-up studies have noted recurrences after both STR and GTR [9, 26, 57, 58, 68, 104, 134]. These tumors have also shown features of malignant transformation and craniospinal dissemination [9, 45, 68, 134]. Rades et al. [95] observed a median time to recurrence for GTR of 36 (range 4–72) months compared with 20 (range 2–72) months for STR. On adding adjuvant RT, it was 39 (24–54) months and 34 (range 2–276) months, respectively [95], thus showing no added RT-related benefit. Others have noted a disease-free survival up to 12.5 years following GTR [62] compared with 18.5 years for STR [68].
Overall, fewer than 100 local recurrences have been reported to date, most of which were associated with atypical neurocytomas, STR, and extraventricular neurocytomas [5, 10–12, 65, 94, 108, 109, 112]. Bertalanffy et al. [9] reviewed 14 CN in patients aged between 16–43 years and equal gender ratio. On follow-up, two patients died postoperatively while one had malignant progression. One who underwent STR had disease progression after 37 months but was stable 10 years after surgery. Five patients had disease recurrence following GTR after a median of 67 (range 51–79) months while the remaining five had a relatively short follow-up period (5–44 months). Histologic and immunohistochemical workup, however, revealed no significant prognostic parameters. The MIB-1 LI ranged between 0.8–11% (mean 4.6%) while that for the malignant transformed tumor was 46.8%. Cases were successfully managed by focal radiotherapy (GKS and interstitial irradiation).
Recurrence in the form of craniospinal dissemination is most unusual, with only seven documented cases. All were lateral ventricle in location, with mean age of 29.9 (range 19–46) years and female preponderance [10, 26, 38, 55, 61, 102, 115]. Cases were managed by GTR (two cases), STR (one case), and STR+RT (four cases). None of these cases showed features of histological atypia, with mean MIB-1 LI of 2% (range 0–4.6%). The interval between surgery and dissemination ranged between 2–36 (mean 18.4) months. Recently, a case of intraventricular neurocytoma with massive brain-stem involvement was reported in a 5-year-old child [66].
Leptomeningeal spread of primary CNS tumors is a well-known phenomenon; however, similar mechanism for primary spinal tumors is a rare [24, 81]. Yamamoto et al. [133] described an unusual neuronal tumor that showed multifocal parenchymal involvement affecting the cerebrum, cerebellum, and spinal cord, with extensive leptomeningeal dissemination at the base of the brain and spinal cord.
Spinocranial dissemination of primary spinal-cord tumor is extremely rare, with limited case reports confined to primitive neuroectodermal tumor (PNET) [64], diffuse astrocytomas [7, 100], germinoma [118], and myxopapillary ependymomas [28] only. Recently, Sharma et al. [109] described a case of recurrent primary atypical spinal neurocytoma, which on follow-up was found to have spinocranial dissemination with evidence of metastasis into the cerebellar vermis.
Predictive factors
The role of histologic atypia and proliferation potential has been extensively investigated in determining the biological behavior of CN. Soylemezoglu et al. [112] concluded that a MIB-1 LI of 2% might be critical in determining recurrence. After a follow-up of 150 months, they observed that patients with MIB-1 LI <2% had a relapse rate of 22% compared with 63% when it was >2%. They also noted a significant correlation between MIB-1 LI with microvascular proliferation (p=0.0006). Sharma et al. [107] observed an elevated MIB-1 LI level in their series, which also had accompanying histologic features of atypia but lacked clinical evidence of poor outcome. However, in the series of Mackenzie et al. [68], MIB-1 LI >2% heralded poor outcome in the form of symptomatic relapse/death, with only one case showing histologic atypia, which was in contrast to cases that had histologic atypia and MIB-1 LI <2% without any relapse. Currently, MIB-1 LI appears to be the best predictor of proliferative potential and clinical outcome of CN [5, 19, 97, 107].
Indian scenario
A study at the All India Institute of Medical Sciences (AIIMS), New Delhi, revealed 49 cases of CN, accounting for 0.39% of all intracranial tumors diagnosed during the 24-year study period (1980–2003). It was observed that cases had a lower mean age (23.7 years) at diagnosis (range 10–42 years), male preponderance (M:F=1.8:1), and higher incidence of involvement of the lateral ventricle (44 cases) in contrast to reports in the Western literature [13, 31, 50]. Out of these 49 cases, 20 CN diagnosed between 1980–1995 constituted material for detailed evaluation and were subsequently published in the literature [107, 108] (Table 4). A major aspect highlighted by these studies [107, 108] was that 11/20 CN had a striking resemblance to oligodendrogliomas and were initially diagnosed as such. Diagnosis of CN was later established by immunohistochemistry and/or electron microscopy on reevaluation. Thirteen of these cases were histologically benign (Group 1) while seven showed mitoses and necrosis (Group 2) [107].
Treatment consisted of surgical resection (total 14 cases, subtotal six), followed by radiotherapy. Except for five cases that succumbed to complications of surgery, the remaining 15 were alive and apparently asymptomatic during the follow-up period, which ranged from 12–72 (mean 32) months. Proliferation index was assessed in formalin-fixed paraffin-embedded tissue in 17 cases using the AgNOR technique and immunohistochemical staining for proliferating cell nuclear antigen (PCNA-PC10 antibody) and Ki-67 antigen (MIB-1 monoclonal antibody). AgNOR counts ranged from 1.2 to 2.6 (mean 1.9±0.4), PCNA labeling index from 0.1 to 5.5 (mean 2.5±1.8), and MIB-1 LI from 0.1 to 3 (mean 0.8±0.02). There was no significant difference in any of these proliferative parameters within histological groups although MIB-1 LI tended to be higher in Group 2 tumors. Further, there was no significant correlation between these proliferative indices and tumor mitotic rates, as well as patient survival.
Recently, we reported a rare case of recurrent primary neurocytoma of the spinal cord in a 24-year-old man [109] who presented with weakness and numbness of the left upper and lower limbs of 2-months duration and a history of previous spinal surgery 10 months previously. Imaging revealed a contrast-enhancing intramedullary mass involving the C5–T1 region. Histologic and immunohistochemical features of both resections were that of atypical neurocytoma, showing occasional mitoses and focal mild vascular proliferation in both specimens, with necrosis being restricted to the initial biopsy only. Marked proliferation with MIB-1 LI of 9% and 10% were noted in the initial and recurrent specimens, respectively. Follow-up showed intracranial dissemination involving the cerebellar vermis, for which the patient underwent surgical resection followed by RT. Postoperative course was uneventful, and the patient improved in terms of cerebellar signs and symptoms. He was alive and on regular follow-up as of 9 months following the procedure.
A retrospective evaluation of intraventricular tumors from a referral center in south India [63] identified eight patients who had undergone radiation therapy following stereotactic biopsy. Seven of these cases were initially diagnosed as oligodendroglioma or ependymoma but were subsequently diagnosed as neurocytoma on synaptophysin immunohistochemistry.
Conclusion
Generally, CN is characterized by a bland benign histology, with an excellent prognosis. Similar histologic picture and immunoprofile also characterizes primary EVN and primary SN [109], with minor variations, the significance of which in terms of biologic behavior remain unclear [37, 59, 85, 120]. In contrast, atypical neurocytomas featuring increased proliferative activity (MIB-1 LI >2%) with or without features of mitoses, necrosis, or microvascular hyperplasia, have been consistently associated with increased risk of recurrence [5, 26, 57, 77, 107, 112].
Safe maximal surgical resection has been associated with a significantly better local control and 5-year survival compared with incomplete resection. The role of radiotherapy is primarily as an adjuvant following surgery, the advantage of which has been noted after STR, especially in patients of atypical neurocytomas. Trial of chemotherapy has been conducted with encouraging results, especially in inoperable cases that have already been irradiated. Currently, CN appear to have a higher incidence of recurrence during long-term follow-up than previously reported, even after complete resection, which have been variably managed by focused radiosurgery. Owing to this, cases need to be regularly followed-up using neuroimaging techniques, even several years after resection.
References
Agranovich AL, Ang LC, Fryer CJH (1993) Central neurocytoma: report of 2 cases and literature review. J Neurooncol 16:47–53
Aker FV, Ozkara S, Eren P, Peker O, Armagan S, Hakan T (2005) Cerebellar liponeurocytoma/lipidized medulloblastoma. J Neurooncol 71:53–59
Anderson RC, Elder JB, Parsa AT, Issacson SR, Sisti MB (2001) Radiosurgery for the treatment of recurrent central neurocytomas. Neurosurg 48:1231–1237
Ando K, Ishikura R, Morikawa T, Nakao N, Ikeda J, Matsumoto T, Arita N (2002) Central neurocytoma with craniospinal dissemination. Magn Reson Med Sci 1:179–182
Ashkan K, Casey AT, D’Arrigo C, Harkness WF, Thomas DG (2000) Benign central neurocytoma. Cancer 89:1111–1120
Barbosa MD, Balsitis M, Jaspan T, Lowe J (1990) Intraventricular neurocytoma: a clinical and pathological study of three cases and review of the literature. Neurosurg 26:1045–1054
Bell WO, Packer RJ, Seigel KR, Rorke LB, Sutton LN, Bruce DA, Schut L (1988) Leptomeningeal spread of intramedullary spinal cord tumors. Report of three cases. J Neurosurg 69:295–300
Bertalanffy A, Roessler K, Dietrich W, Aichholzer M, Prayer D, Ertl A, Kitz K (2001) Gamma knife radiosurgery of recurrent central neurocytomas: a preliminary report. J Neurol Neurosurg Psychiatry 70:489–493
Bertalanffy A, Roessler K, Koperek O, Gelpi E, Prayer D, Knosp E (2005) Recurrent central neurocytomas. Cancer 104:135–142
Brandes AA, Amista P, Gardiman M, Volpin L, Danieli D, Guglielmi B, Carollo C, Pinna G, Turazzi S, Monfardini S (2000) Chemotherapy in patients with recurrent and progressive central neurocytoma. Cancer 88:169–174
Brat DJ, Scheithauer BW, Eberhart CG, Burger PC (2002) Extraventricular neurocytomas. Pathologic features and clinical outcome. Am J Surg Pathol 25:1252–1260
Brown DM, Karlovits S, Lee LH, Kim K, Rothfus WE, Brown HG (2001) Management of neurocytomas: case report and review of the literature. Am J Clin Oncol 24:272–278
Burger PC, Scheirthauer BW, Vogel FS (2002) Neuronal, glioneuronal and neurocytic tumors. In: Burger PC, Scheirthauer BW, Vogel FS (eds) Surgiocal pathology of the nervous system and its coverings, 4th edn. Churchill Livingstone, New York, pp 264–291
Buxton N, Ho KJ, Macarthur D, Vloeberghs M, Punt J, Robertson I (2001) Neuroendoscopic third ventriculostomy for hydrocephalus in adults: report of a single unit’s experience with 63 cases. Surg Neurol 55:74–78
Cerda-Nicholas M, Lopez-Gines C, Peydro-Olaya A, Llombart-Bosch A (1993) Central neurocytoma: a cytogenetic case study. Cancer Genetics Cytogenet 65:173–174
Chuang MT, Lin WC, Tsai HY, Liu GC, Hu SW, Chiang IC (2005) 3-T proton magnetic resonance spectroscopy of central neurocytoma: 3 case reports and review of the literature. J Comput Assist Tomogr 29:683–688
Cobery ST, Noren G, Friehs GM, Chougule P, Zheng Z, Epstein MH, Taylor W (2001) Gamma knife surgery for treatment of central neurocytomas. Report of four cases. J Neurosurg 94:327–330
Coca S, Moreno M, Martos JA, Rodriguez J, Barcena A, Vaquero J (1994) Neurocytoma of spinal cord. Acta Neuropathol (Berl) 87:537–540
Christov C, Alde-Biassette H, Le Guerinel C (1999) Recurrent central neurocytoma with marked increase in MIB-1 labelling index. Br J Neurosurg 13:496–499
Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER Jr, Vedrenne C (1988) Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurg 23:545–556
Daumas-Duport C, Varlet P, Bacha S, Beuvon F, Cervera-Pierot P, Chodkiewiecz JP (1999) Dysembryoplastic neuroepithelial tumors: nonspecific histological forms-a study of 40 cases. J Neurooncol 41:267–280
Daumas-Duport C (1993) Dysembryoplastic neuroepithelial tumors. Brain Pathol 3:283–295
Dodds D, Nonis J, Mehta M, Rampling R (1997) Central neurocytoma: a clinical study of response to chemotherapy. J Neurooncol 34:279–283
Eade OE, Urich H (1971) Metastasising gliomas in young subjects. J Pathol 103:245–256
Ellison DW, Zygmunt SC, Weller RO (1993) Neurocytoma/lipoma (neurolipocytoma) of the cerebellum. Neuropathol Appl Neurobiol 19:95–98
Eng DY, DeMonte F, Ginsberg L, Fuller GN, Jaeckle K (1997) Craniospinal dissemination of central neurocytoma. Report of two cases. J Neurosurg 86:547–552
Englund C, Alvord EC Jr, Folkerth RD, Silbergeld D, Born DE, Small R, Hevner RF (2005) NeuN expression correlates with reduced mitotic index of neoplastic cells in central neurocytomas. Neuropathol Appl Neurobiol 31:429–438
Fassett DR, Pingree J, Kestle JR (2005) The high incidence of tumor dissemination in myxopapillary ependymoma in pediatric patients. Report of five cases and review of the literature. J Neurosurg 102:59–64
Ferreol E, Sawaya R, de Courten-Myers GM (1989) Primary cerebral neuroblastomas (neurocytoma) in adults. J Neurooncol 7:121–128
Figarella-Branger D, Pellissier JF, Daumas D, Delisle MB, Pasquier B, Parent M, Gambarelli D, Rougon D, Hassoun J (1992) Central neurocytomas: critical evaluation of a small-cell neuronal tumor. Am J Surg Pathol 16:97–109
Figarella-Branger D, Soylemezoglu F, Kleihues P, Hassoun J (2000) Central neurocytoma. In: Kleihues P, Cavenee WK (eds) Pathology and genetics of tumors of the nervous system. IARC Press, Lyon, pp 107–109
Friedrichs N, Vorreuther R, Fischer HP, Wiestler OD, Buettner R (2003) Neurocytoma arising in the pelvis. Virchows Arch 443:217–219
Fujimaki T, Matsuno A, Sakai T, Toyoda T, Matsuura R, Ogai M, Kitnaka C, Asai A, Matsutani M, Kirino T (1997) Proliferative activity of central neurocytoma: measurement of tumor volume doubling time. MIB-1 staining index and bromodeoxyuridine labeling index. J Neurooncol 32:103–109
Fujisawa H, Marukawa K, Hasegawa M, Tohma Y, Hayashi Y, Uchiyama N, Tachibana O, Yamashita J (2002) Genetic differences between neurocytoma and dysembryoplastic neuroepithelial tumor and oligodendroglial tumors. J Neurosurg 97:1350–1355
Funato H, Inoshita N, Okeda R, Yamamoto S, Aovagi M (1997) Cystic ganglioneurocytoma outside the ventricular region. Acta Neuropathol (Berl) 94:95–98
Georgen SK, Gonzales MF, McLean CA (1992) Intraventricular neurocytoma: radiologic features and review of the literature. Radiology 182:787–792
Giangespero F, Cenaccchi G, Losi L, Cerasoli S, Bisceglia M, Burger PC (1997) Extraventricular neoplasms with neurocytic features. A clinicopathological feature of 11 cases. Am J Surg Pathol 21:206–212
Goergen SK, Gonzales MF, McLean CA (1992) Intraventricular neurocytoma: radiological features and review of the literature. Radiology 182:787–792
Goto S, Nagahiro S, Ushio Y, Kitaoka M, Nishio S, Fukui M (1993) Immunocytochemical detection of calcineurin and microtubule-associated protein 2 in central neurocytoma. J Neurooncol 16:19–24
Gultekin SH, Dalmau J, Graus Y, Posner JB, Rosenblum MK (1998) Anti-Hu immunolabeling as an index of neuronal differentiation in human brain tumors: a study of 112 central neuroepithelial neoplasms. Am J Surg Pathol 1998 22:195–200
Hamilton R (1997) Case of the month. August 1996-frontal lobe tumor in 11 year old girl. Brain Pathol 7:713–714
Hara A, Araki Y, Shinoda J, Hirayama H, Nikawa S, Sakai N, Yamada H (1993) Central neurocytoma: proliferative assessment by nucleolar organizer region staining. Surg Neurol 39:343–347
Hara M, Aoyagi M, Yamamoto M, Maehara T, Takada Y, Nojiri T, Ohno K (2003) Rapid shrinkage of remnant central neurocytoma after gamma knife radiosurgery: a case report. J Neurooncol 62:269–273
Harris BT, Horoupian DS (2000) Spinal cord glioneuronal tumor with “rosetted” neuropil islands and meningeal dissemination: a case report. Acta Neuropathol 100:575–579
Hassoun J, Gambarelli D, Grisoli F, Pellet W, Salamon G, Pellissier JF, Toga M (1982) Central neurocytoma. An electron microscopic study of two cases. Acta Neuropathol (Berl) 56:151–156
Hassoun J, Soylemezoglu F, Gambarelli D, Figarella-Branger D, von Ammon K, Kleihues P (1993) Central neurocytoma: a synopsis of clinical and histological features. Brain Pathol 3:297–306
Hessler RB, Lopes MB, Frankfurter A, Reidy J, vandenBerg SR (1992) Cytoskeletal immunohistochemistry of central neurocytomas. Am J Surg Pathol 16:1031–1038
Hirschowitz L, Ansari A, Cahill DJ, Bamford DS, Love S (1997) Central neurocytoma arising within a mature cystic teratoma of the ovary. Int J Gynaecol Pathol 16:176–179
Horoupian DS, Shuster DL, Kaarsoo-Herrick M, Shuer LM (1997) Central neurocytoma: one associated with a fourth ventricular PNET/medulloblastomas and the second mixed with adipose tissue. Hum Pathol 28:1111–1114
Ironside JW, Moss TH, Louis DN, Lowe JS, Weller RO (2002) Neuronal and mixed neuronal-glial tumors. In: Ironside JW, Moss TH, Louis DN, Lowe JS, Weller RO (eds) Diagnostic pathology of nervous system tumors. Churchill Livingstone, London, pp 217–252
Ishiuchi S, Tamura M (1997) Central neurocytoma: an immunohistochemical, ultrastructural and cell culture study. Acta Neuropathol (Berl) 94:425–435
Ishiuchi S, Nakazato Y, Iino M, Ozawa S, Tamura M, Ohye C (1998) In vitro neuronal and glial production and differentiation of human central neurocytoma cells. J Neurosci Res 51:526–535
Javedan SP, Manwaring K, Smith KA (2003) Treatment of posterior third ventricular central neurocytoma with endoscopic biopsy, endoscopic third ventriculostomy and stereotactic radiosurgery. Minim Invasive Neurosurg 46:165–168
Jay V, Edwards V, Hoving E, Rutka J, Becker L, Zielenska M, Teshima I (1999) Central neurocytoma: morphological, flow cytometric, polymerase chain reaction, fluorescent in situ hybridization, and karyotypic analyses. Case report. J Neurosurg 90:348–354
Jayasunder R, Shah T, Vaishya S, Singh VP, Sarkar C (2003) In vivo and in vitro MR spectroscopic profile of central neurocytomas. J Magn Reson Imaging 17:256–260
Kawashima M, Suzuki SO, Doh-ura K, Iwaki T (2000) alpha-Synuclein is expressed in a variety of brain tumors showing neuronal differentiation. Acta Neuropathol 99:154–160
Kim DG, Kim JS, Chi JG, Park SH, Jung HW, Choi KS, Han DH (1996) Central neurocytomas: proliferative potential and biological behaviour. J Neurosurg 84:742–747
Kim DG, Paek SH, Kim IH, Chi JG, Jung HW, Han DH, Choi KS, Cho BK (1997) Central neurocytoma: the role of radiation therapy and long term outcome. Cancer 79:1995–2002
Kim DH, Suh YL (1997) Pseudopapillary neurocytoma of temporal lobe with glial differentiation. Acta Neuropathol (Berl) 94:187–191
Kim DG, Chi JG, Park SH, Chang KH, Lee SH, Jung HW, Kim HJ, Cho BK, Choi KS, Han DH (1992) Intraventricular neurocytoma: clinicopathological analysis of seven cases. J Neurosurg 76:759–765
Kim DG, Choe WJ, Chang KH Song IC, Han MH, Jung HW, Cho BK (2000) In vivo proton magnetic resonance spectroscopy of central neurocytomas. Neurosurg 46:329–333
Kubota T, Hayashi M, Kawano H, Kabuto M, Sato K, Ishise J, Kawamoto K, Shirataki K, Iizuka H, Tsunoda S, et al (1991) Central neurocytoma: immunohistochemical and ultrastructural study. Acta Neuropathol (Berl) 81:418–427
Kulkarni V, Rajshekhar V, Haran RP, Chandi SM (2002) Long-term outcome in patients with central neurocytoma following stereotactic biopsy and radiation therapy. Br J Neurosurg 16:126–132
Kwon OK, Wang KC, Kim CJ, Kim IO, Chi JG, Cho BK (1996) Primary intramedullary spinal cord primitive neuroectodermal tumor with intracranial seeding in an infant. Childs Nerv Syst 12:633–636
Lee J, Chang SM, McDermott MW, Parsa AT (2003) Intraventricular neurocytomas. Neurosurg Clin N Am 14:483–508
Lenzi J, Salvati M, Frati A, Raco A, Pichierri A, Giangaspero F, Delfini R (2005) Intraventricular neurocytoma with massive brain stem involvement in a 5-year-old child. Childs Nerv Syst [Epub ahead of print]
Luxton G, Petrovich Z, Jozsef G, Nedzi LA, Apuzzo MI (1992) Stereotactic radiosurgery: principles and comparison of treatment methods. Neurosurg 32:241–259
Mackenzie IR (1999) Central neurocytoma: histologic atypia, proliferation potential, and clinical outcome. Cancer 85:1606–1661
Maiuri F, Spaziante R, De Caro ML, Cappabianca P, Giamundo A, Iaconnetta G (1995) Central neurocytoma: clinico-pathological study of 5 cases and review of the literature. Clin Neurol Neurosurg 97:219–228
Martin JM, Katati M, Lopez E, Bullejos JA, Arregui G, Busquier H, Minguez A, Olivares G, Hernandez V, Arjona V (2003) Linear accelerator radiosurgery in treatment of central neurocytomas. Acta Neurochir (Wien) 145:749–754
Martin AJ, Sharr MM, Teddy PJ, Gardner BP, Robinson SFD (2002) Neurocytoma of the thoracic spinal cord. Acta Neurochir 144:823–828
Maruyama I, Sadato N, Waki A, Tsuchida T, Yoshida M, Fijibayashi Y,Ishii Y, Kubota T, Yonekura Y (1999) Hyperacute changes in glucose metabolism of brain tumors after stereotactic radiosurgery: a PET study. J Nucl Med 40:1085–1090
Megdiche Bazarbacha H, Nagi S, Zouauoi W, Belghith L, Sebai R, Touibi S (2005) Cerebellar liponeurocytoma. Case report. Tunis Med 83:120–122
Metcalf C, Mele EM, McAllister I (1993) Neurocytoma of the retina. Br J Ophthalmol 77:382–384
Miller DC, Koslow M, Budzilovich GN, Burstein DE (1990) Synaptophysin: a sensitive and specific marker for ganglion cells in central nervous system neoplasms. Hum Pathol 21:271–276
Moussa R, Abadjian G, Nader M, Rizk T, Samaha E, Nohra G, Checrallah A, Okias N (2004) Central neurocytoma. Four patients. Neurochirurgie 50:639–646
Mrak RE (1994) Malignant neurocytic tumor. Hum Pathol 25:747–752
Mut M, Guler-Tezel G, Lopes MB, Bilginer B, Ziyal I, Ozcan OE (2005) Challenging diagnosis: oligodendroglioma versus extraventricular neurocytoma. Clin Neuropathol 24:225–229
Nakagawa K, Aoki Y, Sakata K, Sasaki Y, Matsutani M, Akanuma A (1993) Radiation therapy of well-differentiated neuroblastoma and central neurocytoma. Cancer 72:1350–1355
Namiki J, Nakatsukasa M, Murase I, Yamazaki K (1998) Central neurocytoma presenting with intratumoral haemorrhage 15 years after initial treatment by partial removal and irradiations. Neurol Med Chir (Tokyo) 38:278–282
Newman RP, Schaefer EJ, Thomas CB, Oldfield EH (1984) Abetalipoproteinemia and metastatic spinal cord glioblastoma. Arch Neurol 41:554–556
Ng P, Soo YS, Chaseling R, O’Neil P (1996) Intraventricular neurocytoma. Australis Radiol 40:125–133
Nishio S, Morioka T, Suzuki S, Fikui M (2002) Tumors around the Foramen of Monro: clinical and neuroimaging features and their differential diagnosis. J Clin Neurosci 9:137–141
Nishio S, Takeshita I, Fukui M (1990) Primary cerebral ganglioneurocytoma in adult. Cancer 66:358–362
Nishio S, Takeshita I, Kaneko Y, Fukui M (1992) Cerebral neurocytoma: a new subset of benign neuronal tumors. Cancer 70:529–537
Ohgaki H, Eibl RH, Schwab M, Reichel MB, Mariani L, Gehring M, Petersen I, Holi T, Wiestler OD, Kleihues P (1993) Mutations of the p53 tumor suppressor gene in neoplasms of the human nervous system. Mol Carcinog 8:74–78
Ojeda VJ, Jacobsen PF, Papadimitriou JM (1980) Primary cerebral neuroblastomas. Case report with light microscopy, tissue culture and electron microscopy study. Pathology 12:269–274
Pal L, Santosh V, Gayathri N, Das S, Das BS, Jayakumar PN, Shankar SK (1998) Neurocytoma/rhabdomyoma (myoneurocytoma) of the cerebellum. Acta Neuropathol (Berl) 95:318–323
Pearl GS, Takei Y (1981) Cerebellar ‘neuroblastoma”: nosology as it relates to medulloblastomas. Cancer 47:772–779
Pollock BE, Stafford SL (2001) Stereotactic radiosurgery for recurrent central neurocytoma: case report. Neurosurg 48:441–443
Quinn B (1998) Synaptophysin staining in normal brain: importance for diagnosis of ganglioglioma. Am J Surg Pathol 22:550–556
Rabinowicz AL, Abrey LE, Hinton DR, Couldwell WT (1995) Cerebral neurocytoma: an unusual cause of refractory epilepsy. Case report and review of the literature. Epilepsia 36:1237–1240
Rades D, Fehlauer F, Lamszus K, Schild SE, Hagel C, Westphal M, Alberti W (2005) Well-differentiated neurocytoma: what is the best available treatment? Neuro-oncol 7:77–83
Rades D, Fehlauer F, Schild SE (2004) Treatment of atypical neurocytomas. Cancer 100:814–817
Rades D, Fehlauer F (2002) Treatment options for central neurocytoma. Neurology 59:1268–1270
Rades D, Schild SE, Fehlauer F (2004) Defining the best available treatment for neurocytomas in children. Cancer 101:2629–2632
Rades D, Schild SE, Fehlauer F (2004) Prognostic value of the MIB-1 labeling index for central neurocytomas. Neurology 62:987–989
Rades D, Schild SE, Ikezaki K, Fehlauer F (2003) Defining the optimal dose of radiation after incomplete resection of central neurocytomas. Int J Radiat Oncol Biol Phys 55:373–377
Robbins P, Segal A, Narula S, Stoke B, Lee M, Thomas W, Caterina P, Sinclair, Spagnolo D (1995) Central neurocytoma. A clinicopathological, immunohistochemic and ultrastructural study of 7 cases. Pathol Res Pract 191:100–111
Sarabia M, Millan JM, Escudero L, Cabello A, Lobato RD (1986) Intracranial seeding from an intramedullary malignant astrocytoma. Surg Neurol 26:573–576
Schild SE, Scheithauer BW, Haddock MG, Schiff D, Burger PC, Wong WW, Lyons MK (1997) Central neurocytomas. Cancer 79:790–795
Schmidt MH, Gottfried ON, von Koch CS, Chang SM, McDermott MW (2004) Central neurocytoma: a review. J Neurooncol 66:377–384
Schweitzer JB, Davies KG (1997) Differentiating central neurocytoma. Case report. J Neurosurg 86:543–546
Sgouros S, Carey M, Aluwihare N, Barber P, Jackowski A (1998) Central neurocytoma: a correlative clinicopathologic and radiologic analysis. Surg Neurol 49:197–204
Sgouros S, Jackowski A, Carey MP (1994) Central neurocytoma without intraventricular extension. Surg Neurol 42:335–339
Sgouros S, Walsh AR, Barber P (1994) Central neurocytoma of thalamic origin. Br J Neurosurg 8:373–376
Sharma MC, Rathore A, Karak AK, Sarkar C (1998) A study of proliferative markers in central neurocytoma. Pathology 30:355–359
Sharma MC, Sarkar C, Karak AK, Gaikwad S, Mahapatra AK, Mehta VS (1999) Intraventricular neurocytoma: a clinicopathological study of 20 cases with review of the literature. J Clin Neurosci 6:319–323
Sharma S, Sarkar C, Gaikwad S, Suri A, Sharma MC (2005) Primary neurocytoma of the spinal cord: a case report and review of literature. J Neurooncol 74:47–52
Smoker WR, Townsend JJ, Richman MV (1991) Neurocytoma accompanied by Intraventricular haemorrhage: case report and literature review. AJNR Am J Neuroradiol 12:765–770
Soontornniyokkij V, Schelper RI (1996) Pontine neurocytoma. J Clin Pathol 49:764–765
Soylemezoglu F, Schiethauer BW, Esteve J, Kleihues P (1997) Atypical central neurocytoma. J Neuropathol Exp Neurol 56:551–556
Stapleton SR, David KM, Harkness WFJ, Harding BN (1997) Central neurocytoma of the cervical spinal cord. J Neurology, Neurosurgery and Psychiatry 63:119–122
Stephan CL, Kepes JJ, Arnold P, Green KD, Chamberlin F (1999) Neurocytoma of the cauda equina. Case report. J Neurosurg 90:247–251
Takao H, Nakagawa K, Ohtomo K (2003) Central neurocytoma with craniospinal dissemination. J Neurooncol 61:255–259
Taruscio D, Danesi R, Montaldi A, Cerasoli S, Cenacchi G, Giangaspero F (1997) Non random gain of chromosome 7 in central neurocytoma: a chromosomal analysis and fluorescent in situ hybridization study. Virchows Arch 430:47–51
Tatter SB, Borges LF, Louis DN (1994) Central neurocytomas of the cervical spinal cord. Report of two cases. J Neurosurg 81:288–293
Tekkok IH, Sav A (2005) Aggressive spinal germinoma with ascending metastases. J Neurooncol [Epub ahead of print]
Tong CY, Ng HK, Pang JC Ng HK, Pang JC, Hu J, Hui AB, Poon WS (2000) Central neurocytomas are genetically distinct from oligodendrogliomas and neuroblastomas. Histopathol 37:160–165
Tortori-Donati P, Fondelli MP, Rossi A, Fondelli MP, Rossi A, Cama A, Brisigotti M, Pellicano G et al (1999) Extraventricular neurocytoma with ganglionic differentiation associated with complex partial seizures. AJNR Am J Neuroradiol 20:724–727
Tsuchida T, Matsumoto M, Shirayama Y Imahori T, Kasai H, Kawamoto K (1991) Neuronal and glial characteristics of central neurocytoma: electron microscopical analysis of two cases. Acta Neuropathol 81:418–427
Tyler-Kabara E, Kondziolka D, Flickinger JC, Lunsford LD (2001) Stereotactic radiosurgery for residual neurocytoma. Report of four cases. J Neurosurg 95:879–882
Uro-Coste E, Bousquet P, Arrue P, Delisle MB (1999) Central neurocytoma. Immunohistochemical study: MIB-1, p53 and bcl-2. Report of 5 cases. Arch Anat Cyto Pathol 47:13–18
Utsunomiya A, Uenohara H, Suzuki S, Nishimura S, Nishio A, Arai H, Sakurai Y, Suzuki K (2001) A case of anaplastic astrocytoma arising 8 years after initial treatment by partial resection and irradiation for central neurocytoma. No To Shinkei 53:747–751
Valdueza JM, Westphal M, Vortmeyer A, Muller D, Padberg B, Herrmann HD (1996) Central neurocytoma: clinical, immunohistologic, and biologic findings of a human neuroglial progenitor tumor. Surg Neurol 45:49–56
Vallat-Decouvelaere A, Gauchez P, Varlet P, Delisle M, Popovic M, Boissonnet H, Gigaud M, Mikol J, Hassoun J (2000) So-called malignant and extra-ventricular neurocytomas: reality or wrong diagnosis . A critical review about two overdiagnosed cases. J Neurooncol 48:161–172
Vaquero J, Coca S, Oya S, Del Pozo JM, Martinez R, Arias A (1992) Clinicoapthological experience with intraventricular neurocytomas. J Neurosurg Sci 36:31–38
von Deimling A, Janzer R, Kleihues P, Wiestler OD (1990) Patterns of differentiation in central neurocytoma. An immunohistochemical study of eleven biopsies. Acta Neuropathol (Berl) 79:473–479
von Deimling A, Kleihues P, Saremaslani P, Yasargil MG, Spoerri O, Sudhof TC, Wiestler OD (1991) Histogenesis and differentiation potential of central neurocytomas. Lab Invest 64:585–591
von Koch CS, Schmidt MH, Uyehara-Lock JH, Berger MS, Chang SM (2003) The role of PCV chemotherapy in the treatment of central neurocytoma: illustration of a case and review of the literature. Surg Neurol 60:560–565
Westphal M, Stavrou D, Nausch H, Valdueza JM, Herrmann HD (1994) Human neurocytoma cells in culture show characteristics of astroglial differentiation. J Neurosci Res 38:698–704
Wharton SB, Antoun NM, Macfarlane R, Anderson JR (1998) The natural history of a recurrent central neurocytoma-like tumor [published erratum appears in Clin Neuropathol 1998 17:236]. Clin Neuropathol 17:136–140
Yamamoto T, Komori T, Shibata N, Toyoda C, Kobayashi M. (1996)Multifocal neurocytoma/gangliocytoma with extensive leptomeningeal dissemination in the brain and spinal cord. Am J Surg Pathol 20:363–370
Yasargil MG, von Ammon K, von Deimling A, Valavanis A, Wichmann W, Wiestler OD (1992) Central neurocytoma: histopathological variants and therapeutic approaches. J Neurosurg 76:32–37
Yin XL, Pang JC, Hui AB, Ng HK (2000) Detection of chromosomal imbalances in central neurocytomas by using comparative genomic hybridization. J Neurosurg 93:77–81
You H, Kim YI, Im SY, Suh-Kim H, Paek SH, Park S, Kim DG, Jung H (2005) Immunohistochemical study of central neurocytoma, subependymoma, and subependymal giant cell astrocytoma. J Neurooncol 74:1–8
Acknowledgements
The authors acknowledge the contribution of Dr. Ajay Garg, assistant professor, Department of Neuroradiology, All India Institute of Medical Sciences, New Delhi, in the form of providing the neuroradiology images.
Author information
Authors and Affiliations
Corresponding author
Additional information
Comments
William T. Couldwell, Salt Lake City
Since its initial description in 1982 by Hassoun et al., we have learned much about the natural history of this tumor and its response to surgical and radiation therapy. The study of the molecular genetics of this tumor, on the other hand, remains rudimentary, and further work will undoubtedly reap rewards regarding origin, differential diagnosis, and response to treatment. At the present time, the MIB-1 labeling index appears to be the best predictor of proliferative potential and clinical outcome. The role of chemotherapy for treatment of residual tumor has yet to be defined, but promising early reports are emerging. As noted in the report by Sharma et al., larger treatment cohorts need to be evaluated before chemotherapy is incorporated in the conventional treatment schedule.
Rights and permissions
About this article
Cite this article
Sharma, M.C., Deb, P., Sharma, S. et al. Neurocytoma: a comprehensive review. Neurosurg Rev 29, 270–285 (2006). https://doi.org/10.1007/s10143-006-0030-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10143-006-0030-z