
Abstract
Curd initiation and development are complex traits and highly responsive for different temperature ranges in cauliflower. The present study was aimed to identify QTLs for eight traits associated with curding behaviour in diverse germplasm of Indian cauliflower. For this, 92 genotypes of cauliflower and 2 each of tropical broccoli and cabbage were genotyped through genotyping by sequencing (GBS). It generated ≈302 million reads (9.1226E + 10 bp) and identified 35,381 SNPs, maximum from chromosome 3 (4735) with a mean value of 3981.1 SNPs. Ts/Tv ratio was 1.74, suggesting transition bias. STRUCTURE analysis revealed delta value of K = 4 and four subpopulations and prominence of population admixture. In total, 121 significant SNPs were detected for eight traits, 38 for Delhi (North Indian plain) and 83 for Barapani (North-East India). Twelve QTLs were detected for traits associated with regulation of curd formation and development, five of which were for marketable curd length, curd width, days to 50% curd harvest and marketable curd weight from Delhi region and seven for curd length, curd width, days to 50% curd harvest, gross plant weight, leaf length, marketable/net curd weight and number of leaves per plant for Barapani area of North East India. The SNPs identified will be useful for development of markers for curding-related traits and their use in breeding varieties with wider curding plasticity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cauliflower (Brassica oleracea var. botrytis L.) is a widely preferred vegetable, mainly consumed as a fried or boiled, in soup, curry and as pickle. It contains glucosinolates which have anti-cancer properties and also responsible for aroma, pungent taste and bitter flavour (Faulkner et al. 1998). Cauliflower also supplies antioxidants such as selenium (0.47 µg/100 g) and ascorbic acid (47.14 mg/100 g to strengthen immune system (Longvah et al. 2017). New mutants with colourful genes such as Or for β-carotene (Crisp et al. 1975; Kalia et al. 2017), Pr for anthocyanin (Chiu et al. 2005; Singh et al. 2020) and Gr for chlorophyll (Tan et al. 2020) are adding colour and nutrients in cauliflower for growers income and consumers’ health.
Cauliflower is an important household vegetable crop worldwide and being grown in 1.42 mha area with annual production of 26.50 million tonnes. China (40.5%) and India (33.2%) hold major share in global production of cauliflower (FAOSTAT 2018). In India, cauliflower is 6th largest producing vegetable crop with annual production of 8.67 million tonnes from 0.45 mha area (NHB 2018). Its cultivation is expanding spatially and temporally in non-traditional areas after development of tropical cultivars which form curds at higher temperature (20–30 °C); however, productivity shows wide range from 3.86 to 31.0 MT in Indian states (NHB, 2018) which is attributed to genotype and climatic factors since the curding is a thermo-sensitive phase (Matschegewski et al. 2015).
The edible portion of cauliflower is a tender ‘curd’ which consists of floral meristem and proliferated inflorescence (Carr and Irish, 1997; Nimkar and Korla 2011). It contributes around 45% of the plant weight (Rai and Yadav 2005). Interestingly, the temperature regulates initiation and development of the curd but that is through a group of genes such as BoCAL1, BoAP1, CCE1 and BoREM1 (Smith and King 2000). These genes respond to specific temperature range and this temperature–curding interaction was the base for categorising cauliflower into two major groups in India as European (or snowball) and Indian (or tropical) types (Singh et al. 2018). Both of these are distinct from each other for their evolutionary history and growing requirements (Swarup and Chatterjee 1972). In fact, European types were introduced in India in 1822 which served as a source germplasm for evolution of Indian cauliflower through major and minor mutations. Here, the human efforts head great role in selection of annual flowering, heat tolerance and good curding traits (Seshadri and Chatterjee 1996). Presently, India has diverse set of local and exotic germplasm in cauliflower which form curd at a wide range of temperature (10–30 °C). Indian cauliflower forms marketable curd at relatively higher temperature (12–27 °C) than snowball group (10–16 °C). The Indian type is further categorized into early, mid-early and mid-late maturity groups for their temperature requirements to induce curd formation and development at 20–27 °C, 16–20 °C and 12–16 °C, respectively (Singh and Sharma 2003). Fluctuations in temperature during curd development deteriorate quality of curd due to occurrence of bracting, leafiness, yellowing and loose curds (at higher temperature) and buttoning, fuzziness, riciness and pink colouration (at lower temperature) (Bose et al. 2003). This hypersensitive response of cauliflower to temperature fluctuations is not grower-friendly because negligence on part of cultivar selection may cause huge economic loss.
Genetics of curding traits is well investigated in classical studies in Indian cauliflower by Swarup and Pal (1966), Kumaran (1971), Deshpande (1975) and Singh et al. (1975). However, no attempt was made to understand the complexities of these traits using molecular and genomics approaches despite the fact that these are powerful tools for investigating the complex traits involved in domestication, adaptation and yield of crop plants (Emshwiller and Doyle 1998; Olsen and Schaal 1999; Zhou et al. 1999). Researchers used these marker systems in cauliflower for tracking useful traits such as QTLs for curd-related traits (Lan and Paterson 2000; Zhao et al. 2020), orange colour Or gene (Lu et al. 2006) and green curd colour Gr gene (Tan et al. 2020). The molecular markers and next generation sequencing were found to be effective for mapping complex traits.
The ‘genotyping by sequencing’ (GBS) generates huge genomic information on diverse pool of germplasm and allows for detection of single nucleotide polymorphisms (SNPs) and quantitative trait loci (QTLs) (Crossa et al. 2013). The GBS is also effective approach to generate genomic information in crops having high diversity and large genome size. Since no prior genomic information is required, breeders prefer to use it for deciphering the population structure (Elshire et al. 2011). The GBS has been employed in cauliflower by Hasan et al. (2016) for detection of 176 SNPs for leaf appearance rate and curd initiation in relation to temperature and by Stansell et al. (2018) to investigate population admixture in 85 lines of cauliflower, Chinese kale and broccoli. This approach also proved to be useful to distinguish 192 cauliflower accessions of two genebanks of USDA and IPK (Yousef et al. 2018). The GBS analysis of doubled haploid (DH) population could lead to detect five QTLs for heat tolerance in broccoli (Branham et al. 2018) and 18 QTLs for temperature-regulated curd induction and flowering traits in cauliflower (Matschegewski et al. 2015). It was also employed for loci, controlling leaf appearance rate and curd initiation (Hasan et al. 2016; Rosen et al. 2018). However, most of the molecular-genomics studies in cauliflower are performed in European types and their reproducibility in Indian materials is a matter of investigation.
The complexities of the curding trait can be narrowed down by use of robust DNA markers (Zhu et al. 2018). Association mapping accompanied with next generation technology (NGS) offers great advantage for generating genomic information and identification of useful markers (Singh and Singh 2015). It gives higher mapping resolution, hence more advantages as compared to the family mapping for the identification of QTLs in certain situations (Zhu et al. 2008). Association mapping exploits historical and evolutionary recombination events at the population level to resolve the complex trait variation (Nordborg and Tavar 2002; Risch and Merikangas 1996). This is an easy to perform and cost-effective approach for detection of desirable alleles to use in development of robust markers for target trait(s). Association mapping has been successfully used both for specific traits, namely flowering traits in Arabidopsis and quantitative traits in rice, maize and other crops as reviewed by Gupta et al. (2014), Burghardt et al. (2017) and Verdeprado et al. (2018). Thorwarth et al. (2017) performed association analysis in germplasm from two genebanks (IPK and USA) and identified significant QTLs for curding traits.
Considering the diverse groups and huge economic potential of the curding trait in Indian sub-continent, it was decided to generate information on SNPs and QTLs using the GBS approach. The present study was done to assess population structure of Indian cauliflower and perform association mapping of heat stress–related curding traits for identification of QTLs using genotyping by sequencing and phenotyping data from two distinct locations.
Materials and methods
Plant materials and field experimentation
The diversity panel comprised of 92 cauliflower genotypes (10 commercial varieties, 75 fixed inbred lines, 7 advance breeding lines) and two each of tropical broccoli and cabbage. In cauliflower, 92 genotypes were taken from all four maturity groups, i.e. Early (38), Mid-early (25), Mid-late (16) and Late or Snowball (13) groups (Table S1). These sets are based on temperature requirement for curd initiation and development. Two each of sub-tropical flowering broccoli (DC-Brocco-13 and Delhi Purple Broccoli-1 or DPB-1) and tropical cabbage (PA-1 and PA-2) were also included as related crop references in the ‘association panel’ for sequencing. All lines were planted in three-row system (plants = 30) and three replications in complete randomized block design (RBD) at two locations, namely IARI, New Delhi (28°35′ N, 77° 12′ E, 228.6 m above mean sea level) and at ICAR Research complex for NEH Region, Barapani (25°45′ N, 93°50′ E, 295 m above mean sea level) during 2019–2020. Climatic parameters and trial activities at both sites are given in Fig. S1a–b.
Standard crop practices as detailed by Singh and Sharma (2003) were followed for crop raising at both sites. Days to 50% curd initiation (DCI), days to 50% curd maturity (DCH), number of leaves/plant, leaf length (cm), leaf width (cm), gross plant weight (g), curd traits, namely curd length or polar diameter (cm), curd width or equatorial diameter (cm), marketable curd weight (g) and net curd weight (g) were recorded from five random plants in each plot. Curd traits were observed using digital weighing balance and standard ruler as described by Lan and Paterson (2000) and Thorwarth et al. (2017). Mean values of phenotypic data from five plants were calculated for mean and analysed using SAS software tool for ANOVA and basic statistics. Pearson’s correlation coefficient was calculated for all traits.
Genotyping by sequencing of cauliflower
Genomic DNA isolation was performed in 96 samples using the modified CTAB (cetyl-trimethyl-ammonium bromide) method (Doyle and Doyle 1990). SNP genotyping was performed using a genotyping by sequencing (GBS) approach as per original protocol (Elshire et al. 2011). ApeK1 restriction enzyme was used in GBS process to reduce genome complexity and genotype multiple DNA samples. PCR is performed after digestion to increase fragments pool and then GBS libraries are sequenced using NGS technologies, usually resulting in about 100-bp single-end reads. Sequencing was done by Illumina (4000 or Xten 4 Dye Chemistry) platform by NGB Diagnostics Pvt. Ltd., Noida, Uttar Pradesh, India. Sequencing data were processed for quality check by FastQC (version 0.11.5), and filtering, evaluation, SNP calling and variant calling were done using genome analysis toolkit (GATK) version v3.6 (McKenna et al. 2010). Brassica oleracea var. oleracea draft genome (Liu et al. 2014) was taken as reference genome. Reads were mapped to reference genome using the MEM algorithm of BWA (version 0.7.5). Variant calling was done using GATK (version v3.6). Variant filtering was done using VCFTools based on max-missing (0.2) and minor allele frequency [(maf (0.05)]. Chromosome-wise SNP detection, significant SNP identification for target traits, population admixture analysis and linkage disequilibrium (LD) analyses were performed using STRUCTURE software.
Population genetic analysis
Population structure was performed based on 1745 filtered, single nucleotide polymorphisms (SNPs) using a Bayesian Markov Chain Monte Carlo model (MCMC) implemented in STRUCTURE v2.3.4 (Pritchard et al. 2000). The number of presumed sub-populations (K) was set from 2 to ▲10, and each repeated thrice. For each run, burn-in time and MCMC replication number were both set to 50,000 and 100,000, respectively. A model without admixture and correlated allele frequencies was used. The run having maximum likelihood was used to assign 96 genotypes into groups. The most probable K-value was determined by StructureHarvester (Earl 2012) using the log probability of data [LnP(D)] and delta K (ΔK) based on rate of change in [LnP(D)] between successive K-values. The information obtained through model-based clustering was used for working out marker-trait associations, thereby avoiding the spurious associations. Software programme was used to calculate the average distance (expected heterozygosity) between individuals in same sub-population/cluster and allele-frequency divergence (Net nucleotide distance) among populations.
Association analysis was done by compressed mixed linear model (CMLM) approach in genome wide association and prediction integrated tool (GAPIT) version 3 (Lipka et al. 2012). For this, phenotypic data were included from both Delhi and Barapani centres. STRUCTURE (version 2.3.4) was used to analyse population structure and to create Q-matrix for association analysis (Pritchard et al. 2000). Most significant QTLs were identified by CMLM association result by threshold line in Manhattan plot at Padj = 0.05 threshold and around this marker, a range was searched in LD results for associated chromosomes. Population structure was analysed by two approaches: first, using PCA to search for internal patterns of population structure (covariant P) and second, four ancestral subpopulations (K = 4) were estimated by STRUCTURE (covariant Q). Linkage disequilibrium (LD) LD plot was generated using Tomahawk Software (https://mklarqvist.github.io/tomahawk/r).
Genome-wide associations and QTL mapping
Association between the markers and the traits, namely number of days to 50% curd initiation, days to 50% curd maturity, curd length, curd width, gross plant weight, marketable curd weight (with jacket leaves) and net curd weight (without jacket leaves and base stalk) was worked out by compressed mixed linear model (CMLM) which was done using GAPIT (version 3) software for phenotype data from Delhi and Barapani locations using Structure (version 2.3.4) K4 Q-MATRIX. The most significant QTLs were identified by CMLM association result by threshold line in Manhattan plot at Padj = 0.05 threshold and around this marker, a range was searched in *LD results for associated chromosomes.
Results
Phenotypic analysis of plant and curding traits
Field observations on leaf number per plant and seven curding-related traits from 92 genotypes from New Delhi and Barapani centres exhibited a large phenotypic variation for all the observed traits. Furthermore, significant variation was observed between and within all four maturity groups at both locations (Table 1). Genotypic coefficient of variation (GCV) and phenotypic coefficient variation (PCV) were highest for net curd weight at both the locations, i.e. Delhi (37.18%, 39.14%) and Barapani (77.62%, 79.50%), respectively. Broad-sense heritability (H2) differed strongly between traits, and the highest value was observed for DCI (0.94, 0.99) and DCH (0.94, 0.98). Genetic advance as percent of mean was high for net curd weight (72.75, 156.11), marketable curd weight (70.75, 144.89) and gross plant weight (60.83, 156.21) at Delhi and Barapani, respectively.
Genotypes behaved according to their maturity groups for curd-related traits, the early group genotypes had lower values while that of late/snowball group had higher (Fig. 1a–e). The genotypes took 48.0 to 93.3 days for curd initiation in Delhi condition and 16.7 to 123.0 days in Barapani condition with a mean of 65.41 days and 44.27 days, respectively. The curd initiation was observed to be earliest in Pusa Meghna (48 days) at Delhi location and in CC-14 (16.7 days) at Barapani. Both, however, belonged to early maturity group. The DCH ranged from 60.0 to 120 DAT (mean = 81.41 days) in Delhi and 32.0 to 141.3 days in Barapani. Maximum days were taken by KT-22 (120 days) followed by KT-2 (118 days) and KT-6 (116 days) in Delhi while Pusa Snowball K-1 (141.3 days) was followed by KT-20 (141 days) and KT-6 (138 days) at Barapani. Ascending order of genotypes for DCH was Pusa Meghna (60 days) < DC-33–8 (64.7 days) < Sel-7 (65.3 days) < Early Kunwari (65.7 days) for Delhi condition and DC-310–22 < Himgiri < CC-15 < DC-383 < DC-325 for Barapani centre. Curd length was ranged from 8.2 cm (VV) to 16.1 cm (KT-25) at Delhi while 3.8 cm (DC-3023–2) to 10.0 cm (DC-351aa) at Barapani. Late group genotypes had maximum curd width, i.e. KT-25 (16.6 cm) in Delhi and KT-2 (11.8 cm) in Barapani.

(a–e) Comparing Indian cauliflower maturity groups for mean value of curding-related traits in Delhi and Barapani locations
Gross plant weight was highest in KT-22 (2883.3 g) and Pusa Snowball K-1 (1310.0 g). Marketable curd weight was in the range of 320 to 1416 g and 62.0 to 633.3 g at Delhi and Barapani, respectively. The curd weight was significantly low in early group (394.3 ± 96.9 g) than mid-early (521.9 ± 135.07 g), mid-late (674.24 ± 176.8 g) and late group (871.47 ± 95.75 g) at Delhi centre. However, at Barapani centre, the genotypes of mid-early group had lowest mean weight (89.5 ± 47.8 g) than early (106.2 ± 42.8 g), mid-late (147.7 ± 46.3 g) and late group (368.6 ± 137.1 g). Number of leaves per plant in the genotypes ranged from 12.0 to 26.0 with a grand mean of 18.51 while it was in the range from 4.7 to 12.3 at Barapani. At Delhi centre, maximum number of leaves per plant was recorded in BR-2 (26.0) followed by Pusa Early Synthetic (23.8) and DC-85 (23.8), DC-309 (22.7) and DC-383 (22.3); however, at Barapani, KT-22 (12.3) was followed by KT-13–1 (11.8), KT-17 (11.3) and Sel-113 (11.0) DC-DB-6 (4.7).
Sequence prediction and alignment
The GBS assay of the sequencing of 96-plex ApeKI-digested libraries constructed from 96 genotypes of cauliflower (92), broccoli (2) and cabbage (2) was conducted using an Illumina HiSeq 4000. After primary quality filtering process, it generated approximately ≈302 million reads (9.1226E + 10 bp). Average reads per plant sample were 3.146 million (Table 2). After removing low-quality reads and barcode sequences, 7.3279E + 10 bp of sequence data remained, with an average read length of 260.0 bp. Among 96 samples, lowest reads were observed in Pusa Deepali (2,501,628) and highest in DC 310 (8,380,620) with overall mean value of 3,146,579 (Fig. 2a). About 71.8% of the reads were successfully mapped to the reference 02–12 genome of Brassica oleracea (Liu et al. 2014) (Fig. 2b). The amount of data produced from each plant sample was different, similarly the proportion of mapped and paired reads and unmapped reads also.

(a–b) Distribution of sequencing data for each sample after filtering (a) and mapping ratio of generated data (b)
The physical distribution of SNPs was fairly uniform with only 152 gaps of > 200 kb, and all of these occurred within centromeric and pericentromeric regions (Fig. S2). On average for the entire genome, 80.28 SNPs/Mb were found, and this varied between a lowest of 72.8 SNPs/Mb on chromosome 3 and a highest of 84.58 SNPs/Mb on chromosome 6 (Table 3).
The GBS results of the diversity panel resulted into a total of 35,831 SNPs from all the nine chromosomes. The highest SNPs were found on chromosome 3 (4735) and lowest on chromosome 6 (3348) with a mean value of 3981.1 (Table 3). Proportion of heterozygous was in the range of 0.04 to 1.0 with a mean value of 0.46 (Fig. S3). Major allele frequency was in the range of 0.35 to 0.97 and minor allele frequency ranged from 0.02 to 0.5 with mean value of 0.24 (Fig. S4).
Transition and transversion in SNPs
Transition/transversion (Ts/v) analysis revealed transitions (22,583 allelic sites, 63.03%) were frequent than that of transversion (13,248 allelic sites, 36.97%) (Table 4). The Ts/Tv ratio was 1.704 which is larger than expected ratio of 0.5 suggesting that the transition was biased. In transition, frequencies of CT (11,303) and AG (11,280) were almost similar. The AC (10.83%) had the highest frequency in transversion followed by GT (10.69%), AT (8.94%) and CG (6.51%).
Population structure and genetic relatedness
The pairwise genetic distance among 96 genotypes of Cole crops (cauliflower, broccoli, cabbage) was determined from the 35,831 SNP markers. Neighbour joining tree based on these distances showed that the genotypes formed four divergent groups (Fig. 3). Principal component analysis (PCA) also showed clustering of populations according to their putative subpopulations and diversity among the cauliflower genotypes (Fig. 4).

Neighbour-joining dendrogram showing genetic relatedness among the 96 genotypes of Cole crops (cauliflower, broccoli and cabbage) of the diversity panel based on 35,381 SNP markers. Accessions are colour-coded according to the populations’ substructure assignment to cluster G1, G2, G3 and G4 based on STRUTURE results

(a–b) Principal component analysis of SNPs in Indian cauliflower (a) and delta K plot (b)
Furthermore, population structure assessed K values ranging from 1 to 10 on the entire association panel using high-quality 35,831 SNPs. The value of LnP(D) increased continuously with a significant change when delta K changed from 4 to 5. The most likely delta value of K was 4, and such a partitioning of the population into four subpopulations was consistent with significant delta K values (Fig. S5); therefore, this was in accordance with the neighbour-joining tree (Fig. 5). STRUCTURE analysis by 1745 filtered SNPs assigned 96 genotypes to four main subpopulations denoted as G1, G2, G3 and G4 comprising of 2, 4, 15 and 75 genotypes, respectively (Fig. 6). Ninety genotypes (93.75%) were categorized as admixtures. Linkage disequilibrium (LD) decay varied across different chromosomes from 0.25 to 0.33 at r2<0.3 (Fig. S6). The average LD decay for the entire genome was estimated as 0.045 at r2 < 0.2.

Bayesian analysis of population structure in the cauliflower diversity panel assigning the genotypes to four subpopulations. Each genotype is represented by a vertical bar, which is partitioned into K coloured segments that represent individual’s estimated membership coefficient (Q) to the K clusters (STRUCTURE2.3.4)

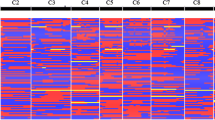
Physical map and chromosomal position of significant QTL (P < 0.01) associated with curding traits in cauliflower. Significant QTL are located on chromosomes C1, C2, C3, C4, C6, C8 and C9 (left: physical position in Mb)
Significant SNP discovery
In total, 121 significant SNPs were identified including 38 from Delhi centre and 83 from that of Barapani region (Table S2). The highest number of significant SNPs was detected for DCI (9) and DCH (9) from Delhi centre for DCI and DCH followed by gross plant weight (16) and marketable curd weight and net curd weight (each 17) for Barapani centre (Table 4). The results pertaining to the GBS for curding traits at two locations are presented in Manhattan plots (Fig. S7).We did not find significant QTL for curd length and curd width from Barapani centre while 2 and 3 were obtained at Delhi centre, respectively. Maximum number of significant SNPs (15SNPs) was obtained on chromosome 4 followed by 6 significant SNPs on chromosome 8. From Barapani centre, the chromosome 4 also had highest number of significant SNPs (33) followed by chromosome 7 (19 SNPs) and chromosome 1 (11 SNPs). In total, 25 SNPs were common at both the locations, highest on chromosome 4 (6 SNPs) followed by 5 SNPs each on chromosome 5 and 8. No common SNPs were detected from chromosome 3 and 6. Among the traits, days to 50% curd initiation had maximum SNPs at both the locations, i.e. 9 at Delhi and 17 at Barapani. Out of these, six SNPs, namely C1_33480103, C4_42620096, C4_42620134, C4_42620145 and C4_42620148 and C4_6974043 were consistent at both the locations and placed on the chromosome C1 and C4. Four SNPs C4_42620096, C4_42620134, C4_42620145 and C4_42620148 were co-localized on chromosome 4. Furthermore, one SNP C5_4851243 was common for marketable curd weight from both locations. However, two significant SNPs SC5_4851243 and SC8_36522162 were common for gross plant weight and marketable curd weight at Delhi location. Two SNPs SC1_34858783 and SC2_18543267 were same for number of leaves per plant and net curd weight for Barapani location (Table 5).
Detection of curding-related QTLs
Twelve QTLs were detected for traits associated with regulation of curd formation and development and the details of these QTLs are given in Table 6 and depicted in Fig. 6. Among them, five QTLs for marketable curd length, curd width, days to 50% curd harvest and marketable curd weight were detected from Delhi region. For Barapani region, seven QTLs were identified for seven traits, namely curd length, curd width, days to 50% curd harvest, gross plant weight, leaf length, marketable/net curd weight and number of leaves per plant located within detected QTL regions. The SNPs for the QTL associated with curd length, curd width and leaf length were C1_33480103, C1_33336570 and C1_33480103, respectively on chromosome 1. The QTL for days to 50% curd harvest, days to 50% curd initiation and marketable curd weight having significant SNP as C2_37720493, C2_48360678 and C4_6974051, respectively, on chromosome 2. The QTL for curd length having an SNP C3_42391757 on chromosome 3, QTL for gross plant weight having a SNP C4_6974051 on chromosome 4, QTL for marketable curd weight and curd width having an SNP C6_31107497 and C6_915168, respectively on chromosome 6 and QTL for days to 50% curd harvest having an SNP C7_20595950 on chromosome 7 have been identified. The genetic linkage between physically neighboured SNPs was assumed at R2 = 0.33 (critical R2 for genetic linkage, based on LD estimation in the cauliflower diversity set).
Discussion
Phylogenetic relationships and population structure
Observations on curding traits from 92 genotypes of cauliflower revealed that the genotypes behaved according to the maturity groups of Indian cauliflower. In cauliflower, maturity groups early, mid-early, mid-late and late/snowball form curds at 20–27 °C, 15–20 °C, 12–16 °C and 10–16 °C, respectively (Singh and Sharma 2003). Curd initiation in early group genotypes, i.e. Pusa Meghna at New Delhi and CC-14 at Barapani was hastened by early occurrence of optimal temperature (i.e. 20–27 °C). Since curd initiation in cauliflower is strictly responsive to temperature, therefore genotypes at both the locations initiated curd formation once they encountered the required range of temperature. Even though the transplanting was almost 1 month delayed at Barapani centre (9th October, 2019) due to continuous rains, the curd initiation in early group genotypes was seen almost in the same week at both the places, i.e. 22nd October, 2019, at Delhi and 25th October, 2019, at Barapani. The mean temperature (15–20 °C) at Barapani centre was lower than Delhi centre (20–25 °C) during October month resulting into low curd weight in the genotypes forming curds in lesser days due to buttoning (formation of small curds due to low temperature).
Indian cauliflower are distinct, but the phylogenetic and population structure analysis revealed that these groups represented by the genotypes having admixture of inter-group segments. This could be due to the breeders’ efforts that the desirable traits for consumers and adaptiveness into different maturity groups were introgressed. This is reflected by the development of varieties/hybrids in Indian cauliflower which grows year-round and form acceptable to very good quality curds (Kalia et al. 2016). Early onset of curd initiation in genotypes of early group Indian cauliflower might be due to change in genomic regions for adaptive trait for sub-tropical climate as revealed by Lin et al. (2018) while highlighting the significance of BoFLC3 and PAN gene in sub-tropical flowering of broccoli. This is because curding is a pre-requisite phase for reproductive phase in cauliflower and there are complex set of genes which respond to prevailing climate.
Genotyping by sequencing analysis
The genotyping by sequencing (GBS) is an effective approach to detect SNPs for mapping the traits of economic interest and for development of robust markers for use in molecular breeding. This is very simple and cost effect approach and is based on high throughput next generation sequencing (Elshire et al. 2011). Estimating the occurrence rate of base substitution correctly is one of the main content in evolution biology studies. In addition, 850 SNPs did not represent the Ts/Tv mutations. Transitions are the most common form of creation of SNPs (Batley et al. 2003) reflecting the high frequency of the C to T mutation following deamination of methylated cytosine residue. In the present study, we could observe that the C to T transition constituted 31.81% of the SNPs observed in diverse set of cauliflower genotypes. Higher number of transition (63.56%) than transversion in present study was in the line of earlier finding of Lopez et al. (2005) wherein they reported 64% transition in cassava. Feltus et al. (2004) reported high level of transition (65.8%) in rice subspecies japonica and indica and they could link it with consequences of epigenetic modifications of CG nucleotide motifs by the DNA methylation.
In association mapping, the power of detection of QTL is low but precision of detection is high as compared to biparental mapping population (Singh and Singh 2015). In present study, we could identify six SNPs, namely C1_33480103, C4_42620096, C4_42620134, C4_42620145 and C4_42620148 and C4_6974043 as common for the traits days to 50% curd initiation and days to 50% curd harvesting at both the locations. Furthermore, one SNP C5_4851243 was common for marketable curd weight for both the locations. However, two SNPs C5_4851243 and C8_36522162 were found to be significant for gross plant weight and marketable curd weight at Delhi location. These significant and common SNPs can be converted to markers for use in simultaneous trait improvement via marker assisted breeding. Only one significant SNP was detected on chromosome 3 which is considered as an important chromosome for Brassica oleracea genome.
Twelve QTLs including five from Delhi region and seven from that of Barapani region were identified for investigated traits. The QTLs from Delhi region for phenotypic traits, namely marketable curd length, curd width, days to 50% curd harvest, days to 50% curd harvest and marketable curd weight on chromosome 1, 2 and 6, respectively and seven QTL from Barapani location for curd length, curd width, days to 50% curd harvest, gross plant weight, leaf length, marketable/net curd weight and number of leaves per plant on chromosome 3, 6, 7, 4, 1, 1 and 2, respectively. Number of leaves per plant also influences the marketable curd weight as observed in correlation analysis (r2= 0.33 for Delhi; r2 = 0.76 for Barapani locations). Significant SNPs for this trait can also be investigated to establish their usefulness. Leaf number is a stable marker of the end of juvenile phase in different light conditions but influenced by planting time and temperature (Hand and Atherton 1987). Booij and Struik (1990) reported that the curd diameter increased slowly at 22 °C than at 14 °C and relationship between the number of leaves and the curd diameter.
Thirteen SNPs were found to be common for both marketable curd weight and net curd weight from Barapani location. This is well justified by strong positive correlation between these traits (r2 = 0.977). These SNPs have potential to explore for development of markers for these traits; however, there is a need to validate the results by having subsequent phenotyping experiment.
QTL detection and usefulness
In the present study, we observed a QTL for marketable curd weight with phenotypic variation explained value of 31.21%. Zhao et al. (2020) reported 20 QTLs having phenotypic variation explained ranging from 7.69 to 25.10%. Among them, four QTLs were for curd architectural traits, namely qSL.C6-1 and qSL.C6-2 for stalk length and qCS.C6-1 and qCS.C6-4 for curd solidarity which were located in the same chromosome region and indicated pleiotropic effect or are tightly linked. In our study, we observed twelve QTLs on 6 chromosomes for various curding-related traits in both the regions. Similar attempts were made by Zhao et al. (2016) and constructed a high genetic map including 2741 SNPs and identified QTLs for curd architectural traits on chromosomes 6. However, we could observe common SNPs for days to 50% curd initiation and 50% curd maturity as well as for marketable curd weight and net curd weight. This could be due to common genetic factors for these traits or there may be a strong linkage or co-localizations of SNPs which was also reported by Zhao et al. (2020) while studying QTLs for curd solidarity on chromosome 6. They also reported two QTLs for basal diameter (qBD.C3-1 and qBD.C3-2) on chromosome 3 and three QTLs for stalk angle including two QTLs (qSA.C1-1 and qSA.C1-2) on chromosome 3 and one on chromosome 6 (qSA.C6-1). It is to mention that the curd solidity is directly related with marketable curd weight (Nieuwhof and Garretsen 1961) and Zhou et al. (2020) reported a QTL qCS.C5 on chromosome 5. In the present study, we also detected a QTL for marketable curd weight on chromosome 6 and 4 for Delhi and Barapani regions, respectively. However, there is need to be investigate the correlation between these two traits.
With respect to heat tolerance in cauliflower, the days taken to curd initiation and marketable curd weight are important indicators, because lower or higher temperature than the desired one during curd induction phase leads to early or delayed in curding, respectively. Temperature during curd development directly affects the curd growth and quality. But, the information on targeted breeding for heat tolerance in crop plants particularly in Cole crops is scarce. Branham et al. (2018) developed 1423 SNPs through GBS and detected five QTLs for heat tolerance in broccoli and one of them had positive epistatic interaction for heat tolerance trait.
We observed two QTLs on chromosome 4 for curd marketable weight and gross plant weight. Matschegewski et al. (2015) also reported two QTLs on chromosome 4 (a significant QTL FT-T1 and an additional FT-T1) for curd induction in response to higher daily temperature (minimum 15.5 °C and maximum 30 °C) using GBS approach. Besides, they also identified 18 other QTLs on C1, C2, C3, C4, C6, C8 and C9 having association with curd initiation in cauliflower. Number of leaves per plant is crucial factor on curd initiation and Lin et al. (2019) reported that the cultivars of tropical cauliflower initiate curd development early than those of snowball group cultivars.
We mapped QTL12 on chromosome 2 for number of leaves per plant with explained phenotypic variation of 31.21%. Hasan et al. (2016) also reported one QTL for final leaf number (LAR) on chromosome 2 with phenotypic variation of 21.8%; however, they could detect 28 QTLs additionally on eight other chromosomes. They reported negative correlations between LAR and DCI and co-localization of QTLs on C4 and C6 which suggest for role of LAR in curd induction.
Williams and Atherton (1990) indicated that curd initiation occurred earlier with fewer leaves at a low temperature (5 °C) and that more leaves were required at a warm temperature (20 °C). The findings are in consistency with previous studies of Zhao et al. (2020) for high heritability of curding traits, showing the feasibility of genetic improvement for desirable curd architecture.
The STRUCTURE analysis revealed existence of four sub-groups (KQ = 4) as G1, G2, G3 and G4 with 2, 4, 15 and 75 genotypes, respectively. Only six genotypes (6.25%) did not show admixture while remaining genotypes had low to high level of admixture. Maximum share of genotypes (39.58%) showed admixture of two subpopulations while 32.29% genotypes had genetic makeup from three subpopulations. Interestingly, 21.87% genotypes had almost balanced admixture from four subpopulations. The admixture was attributed during evolution of Indian cauliflower in past 200 years and as a result of intentional efforts to introgress economic traits in tropical types (Swarup and Chatterjee 1972). Dey et al. (2019) also had similar findings and highlighted the role of frequent introgression from European types for evolution of present day cultivated Indian types.
The present study found strong effect of temperature on determination of curd initiation, curd harvest period and other curding-related traits in the diverse sets of cauliflower. The significant common SNPs identified at two diverse locations can be converted into markers for use in simultaneous improvement of traits via marker assisted breeding. This is the first detailed study on Indian cauliflower and the promising QTLs regions will be helpful for breeders to establish their potential along with already reported candidate genes for curding. Furthermore, this will help to dissect the allelic diversity and to map the genetic variation in curding traits to temperature response in Indian cauliflower. Genomic information on curding traits of Indian cauliflower will facilitate designing robust molecular markers to develop varieties/hybrids with wider adaptive plasticity.
Change history
20 November 2021
A Correction to this paper has been published: https://doi.org/10.1007/s10142-021-00817-5
References
Batley J, Mogg R, Edwards D, O’Sullivan H, Edwards KJ (2003) A high- throughput SNuPE assay for genotyping SNPs in the flanking regions of Zea mays sequence tagged simple sequence repeats. Mol Breed 11:111–120
Booij R, Struik PC (1990) Effects of temperature on leaf and curd initiation in relation to juvenility of cauliflower. Sci Hortic 44:201–214
Bose TK, Som MG, Kabir J (2003) Vegetable crops. NayaProkash, Kolkata, India. pp. 152–156
Branham SE, Stansell ZJ, Couillard DM, Farnham MW (2018) Quantitative trait loci mapping of heat tolerance in broccoli (Brassica oleracea var. italica) using genotyping-by-sequencing. Theor Appl Genet 130:529–553
Burghardt LT, Young ND, Tiffin P (2017) A guide to genome wide association mapping in plants. Curr Protoc Plant Biol 1:22–38
Carr SM, Irish VF (1997) Floral homeotic gene expression defines developmental arrest stages in Brassica oleracea L. var. botrytis and italica. Planta 201:179–188
Chiu LW, Prior RL, Wu X, Li L (2005) Toward identification of the candidate gene controlling anthocyanin accumulation in purple cauliflower (Brassica oleracea L. var. botrytis). American Society of Plant Biologist Annual Meeting, pp. 628 (Abtr.)
Crisp P, Jewell PA, Gray AR (1975) Improved selection against the purple colour defect of cauliflower curds. Euphytica 24:177–180
Crossa J, Beyene Y, Kassan S, Perez P, Hickey JM, Chen C, Jannink JL (2013) Genomic prediction in maize breeding populations with genotyping-by-sequencing. G3: Genes Genomes Genet 3:1903–1926
Deshpande AA (1975) Genetical studies in early cauliflower. M.Sc Thesis, PG School, IARI, New Delhi
Dey SS, Bhatia R, Sharma K, Bhardwaj I, Pandey CD, Parkash C, Kumar R (2019) Frequent introgression of European cauliflowers in the present day cultivated Indian cauliflowers and role of Indian genotypes in the evolution of tropical cauliflower. Euphytica 215:23. https://doi.org/10.1007/s10681-019-2346-0
Doyle J, Doyle JI (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. https://doi.org/10.1371/journal.pone.0019379
Emshwiller E, Doyle JJ (1998) Origins of domestication and polyploidy in oca (Oxalis tuberosa: Oxalidaceae): nrDNA ITS data. Am J Bot 85:975–985
FAOSTAT (2018) Food and Agriculture Organization of United Nations. Italy, Rome
Faulkner K, Mithen R, Williamson G (1998) Selective increase of the potential anticarcinogen 4- methylsulphinylbutyl glucosinolate in broccoli. Carcinogenesis 19:605–609
Feltus FA, Wan J, Schulze SR, Estill JC, Jiang N, Paterson AH (2004) An SNP resource for rice genetics and breeding based on subspecies indica and japonica genome alignments. Genom Res 14:1812–1819
Gupta PK, Kulwal PK, Jaiswal V (2014) Association mapping in crop plants: opportunities and challenges. Adv Genet 85:109–147
Hand DJ, Atherton JG (1987) Curd initiation in the cauliflower: I. Juvenility J Exp Bot 38:2050–2058
Hasan Y, Briggs W, Matschegewski C, Ordon F, Stutzel H, Zetzsche H, Uptmoor R (2016) Quantitative trait loci controlling leaf appearance and curd initiation of cauliflower in relation to temperature. Theor Appl Genet 129:1273–1288
Kalia P, Singh S, Prakash C, Dey SS (2016) Cole vegetables enhancing volume of vegetable basket. Indian Hort 61:77–81
Kalia P, Muthukumar P, Soi S (2017) Marker-assisted introgression of the Or gene for enhancing β-carotene content in Indian cauliflower. ISHS Acta Hort. 1203:121–128. https://doi.org/10.17660/ActaHortic.2018.1203.19
Kumaran NM (1971) Studies on combining ability, gene effects and heterosis in cauliflower, PhD Thesis IARI, New Delhi
Lan TH, Paterson AH (2000) Comparative mapping of quantitative trait loci sculpting the curd of Brassica oleracea. Genetics 155:1927–1954
Lin CY, Chen KS, Chen HP, Lee HI, Hsieh CH (2019) Curd initiation and transformation in tropical cauliflower cultivars under different temperature treatments. HortScience 54:1351–1356
Lin YR, Lee JY, Tseng MC, Lee CY, Shen CH, Wang CS, Hwu KK (2018) Subtropical adaptation of a temperate plant (Brassica oleracea var. italica) utilizes non-vernalization-responsive QTLs. Sci Rep 8:1–10. https://doi.org/10.1038/s41598-018-31987-1
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ et al (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28(18):2397–2399
Liu S, Liu Y, Yang X, Tong C, Edwards D, Parkin IA, Wang X (2014) The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun 5:1–11. https://doi.org/10.1038/ncomms4930
Longvah T, Ananthan R, Bhaskarachary K, Venkaiah K (2017) Indian food composition tables. National Institute of Nutrition Indian (Council of Medical Research), Hyderabad, India
Lopez C, Piegu B, Cooke R, Delseny M, Tohme J, Verdier V (2005) Using cDNA and genomic sequences as tools to develop SNP strategies in cassava (Manihot esculenta Crantz). Theor Appl Genet 110:425–431
Lu S, Van Eck J, Zhou X, Lopez AB, O’Halloran DM, Cosman KM, Kochian LV (2006) The cauliflower Or gene encodes a DnaJ cysteine-rich domain-containing protein that mediates high levels of β-carotene accumulation. Plant Cell 18:3594–3605
Matschegewski C, Zetzsche H, Hasan Y, Leibeguth L, Briggs W, Ordon F, Uptmoor R (2015) Genetic variation of temperature-regulated curd induction in cauliflower: elucidation of floral transition by genome-wide association mapping and gene expression analysis. Front Plant Sci 6:720. https://doi.org/10.3389/fpls.2015.00720
McKenna A, Hanna M, Banks E et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303
NHB (2018) National Horticulture Database. http://nhb.gov.in/statistics/State_Level/2017- 18
Nieuwhof M, Garretsen F (1961) The solidity of the cauliflower curd. Euphytica 10:301–306
Nimkar SA, Korla BN (2011) Evaluation of biparental progenies developed through bud and mixed pollination for horticultural and quality traits in late cauliflower (Brassica oleracea var. botrytis L.). Indian J Agri Res 45:215–220
Nordborg M, Tavar ES (2002) Linkage disequilibrium: what history has to tell us. Trends Genet 18:83–90
Olsen KM, Schaal BA (1999) Evidence on the origin of cassava: phylogeography of Manihot esculenta. Proc Natl Acad Sci USA 96:5586–5591
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rai N, Yadav DS (2005) Advances in vegetable production. Researchco Book Centre, New Delhi. pp. 1–1007
Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517
Rosen A, Hasan Y, Briggs W, Uptmoor R (2018) Genome-based prediction of time to curd induction in cauliflower. Front Plant Sci 9:78. https://doi.org/10.3389/fpls.2018.00078
Seshadri VS, Chatterjee SS (1996) The history and adaptation of some introduced vegetable crops in India. Veg Sci 23:114–140
Singh BD, Singh AK (2015) Marker-assisted plant breeding: principles and practices. Springer, Switzerland AG, pp 77–122
Singh DP, Swarup V, Chattarjee SS (1975) Genetical studies in Indian cauliflowers (Brassica oleracea var. botrytis L.): heterosis and combining ability in maturity group 1. Veg Sci 32:41–46
Singh PK, Singh S, Sharma SR (2018) Tropical Indian cauliflower. In: Peter KV (ed): Genesis and evolution of horticultural crops Vol. II. Astral International Pvt Ltd New Delhi, Pp. 113–123.
Singh R, Sharma SR (2003) Cauliflower. In: Singh N, Thamburaj (eds) Textbook of vegetables tubercrops and spices. ICAR, New Delhi. pp 81–82
Singh S, Kalia P, Meena RK, Mangal M, Islam S, Saha S, Tomar BS (2020) Genetics and expression analysis of anthocyanin accumulation in curd portion of Sicilian purple to facilitate biofortification of Indian cauliflower. Front Plant Sci 10:1766. https://doi.org/10.3389/fpls.2019.01766
Smith LB, King GJ (2000) The distribution of BoCAL-a alleles in Brassica oleracea is consistent with a genetic model for curd development and domestication of the cauliflower. Mol Breed 6:603–613
Stansell Z, Hyma K, Fresnedo-Ramirez J, Sun Q, Mitchell S, Bjorkman T, Hua J (2018) Genotyping-by-sequencing of Brassica oleracea vegetables reveals unique phylogenetic patterns, population structure and domestication footprints. Hort Res 5:1–10. https://doi.org/10.1038/s41438-018-0040-3
Swarup V, Pal AB (1966) Gene effects and heterosis in cauliflower I. Indian J Plant Genet Resour 26:269–281
Swarup V, Chatterjee SS (1972) Origin and genetic improvement of Indian cauliflower. Econ Bot 26:381–393
Tan H, Wang X, Fei Z, Li H, Tadmor Y, Mazourek M, Li L (2020) Genetic mapping of green curd gene Gr in cauliflower. Theor Appl Genet 133:353–364
Thorwarth P, Yousef EAA, Schmid KJ (2017) Genomic prediction and association mapping of curd-related traits in gene bank accessions of cauliflower. G3. Genes Genom Genet 8:707–718
Verdeprado H, Kretzschmar T, Begum H, Raghavan C, Joyce P, Lakshmanan P, Cobb JN, Collard BYC (2018) Association mapping in rice: basic concepts and perspectives for molecular breeding. Plant Prod Sci 3:159–176
Williams CA, Atherton JG (1990) A role for young leaves in vernalization of cauliflower: I. analysis of leaf development during curd induction. Physiol Plant 78:61–66
Yousef EA, Muller T, Borner A, Schmid KJ (2018) Comparative analysis of genetic diversity and differentiation of cauliflower (Brassica oleracea var. botrytis) accessions from two ex situ gene banks. PloS One, 13:e0192062. https://doi.org/10.1371/journal.pone.0192062
Zhao ZQ, Sheng XG, Yu HF, Wang JS, Shen YS, Gu HH (2020) Identification of QTLs associated with curd architecture in cauliflower. BMC Plant Biol 20:1–8. https://doi.org/10.1186/s12870-020-02377-5
Zhao Z, Gu H, Sheng X, Yu H, Wang J, Huang L, Wang D (2016) Genome-wide single-nucleotide polymorphisms discovery and high-density genetic map construction in cauliflower using specific-locus amplified fragment sequencing. Front Plant Sci 7:334. https://doi.org/10.3389/fpls.2016.00334
Zhou X, Jellen EN, Murphy JP (1999) Progenitor germplasm of domesticated hexaploid oat. Crop Sci 39:1208–1214
Zhu S, Zhang X, Liu Q, Luo T, Tang Z, Zhou Y (2018) The genetic diversity and relationships of cauliflower (Brassica oleracea var. botrytis) inbred lines assessed by using SSR markers. PloS One, 13: e0208551. https://doi.org/10.1371/journal.pone.0208551
Zhu X, Van Eck J, Li L (2008) Use of the cauliflower Or gene for improving crop nutritional quality. Biotechnol Annu Rev 14:171–190. https://doi.org/10.1016/s1387-2656(08)00006-9
Acknowledgements
Authors acknowledge the PG School, IARI, for fellowship and NAPEH-CAAST (IARI) for financial support. GBS analysis and CRP-HT for financial support for field evaluation of genotypes. We thank directors, IARI and ICAR-NEH-RC, for facilities at New Delhi and Barapani centres for overall support. We also acknowledge IARI Regional Station, Katrain, Himachal Pradesh, India, for sharing seeds of snowball cauliflower.
Author information
Authors and Affiliations
Contributions
RKN conducted field trials, extracted genomic DNA and drafted manuscript. SS conceived the idea, directly supervised trials, interpreted results and finalized manuscript. VKV supervised trial at ICAR-NEH, Barapani. BBS helped in interpretation of results and review of manuscript. NS helped in interpretation of GBS data. MAI reviewed manuscript. TKB helped in conceptualization, resources and overall guidance for trial.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Originally, the author’s affiliation has been erroneously published. This is now updated.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rakshita, K.N., Singh, S., Verma, V.K. et al. Understanding population structure and detection of QTLs for curding-related traits in Indian cauliflower by genotyping by sequencing analysis. Funct Integr Genomics 21, 679–693 (2021). https://doi.org/10.1007/s10142-021-00811-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-021-00811-x