
Abstract
Wheat-related disorders are well-studied health problems. Knowledge of the composition and amounts of epitopes present in a single wheat sample represents a significant gap, and the detailed wheat proteome datasets now available can provide the necessary information to carry out an estimation of allergen prediction for a single cultivar. The combined use of genome sequence and allergen databases, prediction methodology, and cereal chemistry results in better understanding of the level of toxicity present in the end-products produced from wheat flour. The workflow presented in this review provides information about the number and distribution of epitopes at single protein, or protein fraction, levels. In addition, epitopes present in the highest frequency and harmful proteins expressed in the highest amount can be identified. The “epitope toxicity” value obtained in this way is a significant research output from the analysis of large datasets that can be applied to the food industry.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The availability of extensive data sets defining the entire gene complement of wheat is of particular interest in predicting proteins that may be related to human disorders such as celiac disease (Osorio et al. 2012) and sensitivities to wheat (Sapone et al. 2012; Juhász et al. 2012). Large databases that combine protein sequences per se and sequences obtained from the translation of genes provide a convenient template for analyzing mass spectroscopy-based peptide sequence information (Dupont et al. 2011). Predicted gene translations from the KOMUGI full-length cDNA database (http://www.shigen.nig.ac.jp/wheat/komugi/) are a well-established resource and provide the additional information for tissue expression.
The unusual amino acid composition and primary structure of prolamins in the cereal grain make them a specific focus for defining nutrition- and health-related consequences (Sampson and Metcalfe 1992). Gluten-containing food can cause health problems for a significant number of people who consume foods derived from wheat, rye, barley, and oats (van Heel and West 2006). The spectrum of gluten-related disorders and a new nomenclature to describe them has been provided by Sapone et al. (2012). In Sapone et al. (2012), the suggested nomenclature for wheat gluten-related disorders is divided into three main groups, namely autoimmune, allergy, and not autoimmune-allergic disorders. Two of these disorder types are relatively well-described; however, further studies are needed to reveal mechanisms related to the diverse symptoms of allergic reactions. The autoimmune disorders include celiac disease (CD) (Anderson and Wieser 2006), gluten ataxia, and dermatitis herpetiformis (Laurièrre et al. 2006). The allergic reactions involve respiratory allergy (Amano et al. 1998), food allergy (Mills et al. 2003), wheat-dependent exercise-induced anaphylaxis (Armentia et al. 1990), and contact urticaria. In both the autoimmune and allergic reactions, the T cells are the main mediators. While in celiac disease the characteristic symptoms are developed after the different receptor proteins (for example, HLA DQ) bind to the antigens to the surface of the antigen-presenting cells, the development of wheat allergies is mediated more directly by the recognition of allergens by immunoglobulin E (IgE) epitopes bound to the mast cells (Catassi and Fasano 2008).
Symptoms of celiac disease are triggered by an abnormal response of autoantibodies, such as tissue transglutaminase on proline- and glutamine-rich wheat gluten proteins and their barley and rye homologs (Green and Cellier 2007). Consumption of oat products is considered safe by the majority of CD patients, although sensitive cases have been also detected (Garsed and Scott 2007). Celiac disease shows a strong association with HLA–DQ alleles, and carriage of certain HLA molecules is the most significant genetic factor to develop detrimental symptoms (Sollid et al. 2012), although the importance of differentiating between epitopes targeting different HLA molecules was evident. The presence of HLA DQ molecules such as DQ2.5, DQ8, and DQ2.2 were clear markers for developing autoimmune symptoms, with modifying effects coming from genetic and environmental factors (Anderson et al. 2000). Patients suffering from celiac disease produce a range of autoimmune responses to several alpha gliadin, LMW glutenin, and gamma gliadin peptides in the consumed wheat products. There are a number of different conditions, which need to be realized simultaneously to provoke an autoimmune response (Anderson et al. 2000). Based on the knowledge of genetic, immunological, and biochemical mechanisms of CD, a set of criteria can be defined for the structure of an active CD epitope: (a) a size of nine amino acids, (b) the presence of a tissue transglutaminase 2 (tTG) enzyme-binding site, and (c) being surrounded by amino acids with defined charge and hydrophobicity (Sollid et al. 2012).
In wheat allergy, allergen-presenting B and T cells have the same level of impact and, similar to other food allergies allergic reactions, are the result of specific crosslinks between immunoglobulin E and short peptides rich in glutamine and proline that result from the breakdown of wheat seed proteins by endogenous proteases (Sapone et al. 2012). This interaction stimulates basophils and mast cells to release chemical mediators, such as histamines, resulting in different categories of inflammatory reactions. The allergenic regions of protein recognized by the binding sites of IgE molecules are called IgE-binding epitopes. These epitopes can be classified into two categories, conformational and linear epitopes, and both types are involved in the development of allergic reactions (Akagawa et al. 2007).
Independently from the pathology of the disorder, most of the causative allergens are members of the prolamin superfamily. The prolamin superfamily contains the highest number of allergens of all allergic protein superfamilies (Radauer and Breiteneder 2007). Four of its eight member families, prolamins, nsLTPs, alpha-amylase/protease inhibitors, and 2S albumins contain allergens (Breiteneder and Radauer 2004). The identification of the existence of the prolamin superfamily was based on the discovery of the presence of a conserved cysteine skeleton Cys-Xaa n -Cys-Xaa n -Cys-Cys-Xaa n -Cys-Xaa-C-Xaa n -Cys-Xaa n -Cys (Breiteneder 2008). Characterization of the wheat allergen epitopes bound to IgE antibodies is essential for understanding the mechanisms of wheat allergy-related symptoms as well as for the development of new tools in allergy predictions. Wheat grain proteins that have been proven to interact with IgE include thioredoxin, wheat germ agglutinin, peroxidase, triosephosphate isomerase, serpins, and glyceraldehyde-3-phosphate dehydrogenases (Tatham and Shewry 2008).
Computational tools used in CD/wheat allergy studies
The T- and B- (IgE) epitopes are both relevant targets for allergen identification and prediction, and a number of tools and methods have been developed. The computational and bioinformatic tools used in allergen identification, allergen prediction, and also in vaccine design have been extensively explored and reviewed by several research groups (Brusic et al. 2003; Tomar and De 2010; Moolhuijzen 2011). The tools follow a pipeline that includes sequence analysis and comparison, prediction, and structural analysis (Brusic et al. 2003).
The sequence analysis and comparison tools focus on classification and characterization of potential allergens based on experimentally confirmed epitope sets and homolog allergens identified from related species. There are several datasets of epitopes readily available for epitope mapping and allergen characterization (e.g., www.immunepitope.org; www.allergome.org; http://allergen.nihs.go.jp). In addition, Sollid and coworkers (2012) have documented a collection of core celiac disease epitopes. All of the epitopes are nine amino acids in length, with at least one tissue transglutaminase target place and each epitope assigned to one of the HLA DQ alleles (HLA DQ2., HLA DQ2.2, HLA DQ8, and HLA DQ8.5). Similarly, Tatham and Shewry (2008) provided a detailed epitope list focused on epitopes which could be assigned to wheat-dependent exercise-induced anaphylaxis (WDEIA). The epitope lists are well-annotated and characterized for the CD response (Sollid et al. 2012), but deficiencies exist in the wheat allergy-related epitope lists because it is not known whether the different syndromes classed as wheat allergy can be assigned to any of the major histocompatibility complex classes I or II. Interpretation of the overall outputs thus needs to take into account that, whereas CD is now the best-known human autoimmune disease, the WA is an extremely very diffuse area with conflicting clinical results (Catassi and Fasano (2008). The interpretation of the proteins that come up “positive” for WA epitopes, in particular, therefore need to be treated with caution and to consider the frequency of possible false-positives. A further variable to consider is the limitation of the experimentally known epitope contributing to databases. Currently, the knowledge base is dominated by gluten-based epitopes, and members of the prolamin superfamily, such as serpins and nsLTPs, are not fully represented.
Prediction tools for allergic or celiac potential are based on the special characteristics with respect to either function or composition and chemical structure of proteins. The requirements for B cell or IgE epitope prediction is based on the chemical and structural composition analysis of the potential allergens. The epitope is considered to occur on the surface of the protein and is thus more flexible than the rest of the protein. The epitope region requires a high degree of exposure to the solvent, and the amino acids making up the epitope are generally charged and hydrophilic.
The main methods for the identification of B cell epitopes currently in use are:
-
1.
The Parker hydrophilic prediction method (Parker et al. 1986) utilized three parameters, hydrophilicity, Janin’s scale accessibility, and the flexibility of Karplus and Schultz (1985). The hydrophilicity parameter was calculated using a HPLC-retention co-efficient-based model synthetic peptides studies. The surface profile was determined by summing the parameters for each residue of a seven-residue segment and assigning the sum to the fourth residue. This remains as one of the most useful prediction algorithms (Parker et al. 1986).
-
2.
Surface accessibility prediction (Emini et al. 1985)
-
3.
Beta-turn prediction (Chou and Fasman 2006)
-
4.
Exposed surface, polarity, and antigenic propensity of polypeptide chains have been correlated with the location of continuous epitopes. Kolaskar’s semiempirical method uses physiological properties of amino acid residues and the frequencies of occurrence of amino acids in experimentally known epitopes (Kolaskar and Tongaonkar 1990).
Conditions which determine the capacity of a T cell epitope within a plant protein to cause a response in humans include: the resistance to proteolytic digestion; presence of an at least nine-amino-acid-long core peptide recognized by one of the HLA DQ classes mentioned above; and the presence of at least one glutamine residue as a target for transglutamination carried out by tissue transglutaminase (Sollid et al. 2012). Experimentally determined affinity data have been used to develop a variety of T cell-based prediction methods.
The algorithms used to model the binding of unique HLA alleles to possible allergen peptides include:
-
1.
Motif-based systems, which are straightforward methods of epitope detection; however, it results in a relatively large number of false-positive and false-negative results (Rammensee et al. 1999).
-
2.
Support vector machines (SVMs) statistically separate analyzed peptides into HLA DQ binder and non-binder groups, based on the different HLA DQ alleles (Donnes and Elofsson 2002).
-
3.
Hidden Markov models, which is considered as the most powerful tool for T cell epitope prediction uses statistical models to determine peptides which possess binder-like characteristics (Brusic et al. 2002).
-
4.
Artificial neural networks (ANNs) consider the identity of amino acid residues in a peptide sequence and the interactions between adjacent amino acids in a potential epitope and the HLA DQ molecule (MacNamara et al. 2009). Once an ANN is trained for a particular molecule, it can predict the binding affinity of novel peptide sequences used in MHC-II class epitope prediction (Brusic et al. 1998).
-
5.
Quantitative structure–activity relationship analysis considers the optimized individual residue-to-residue interactions in the HLA DQ groove (Doytchinova et al. 2005).
The use of these T cell epitope prediction methods has been reviewed among others by Brusic et al. (2004) and Davies et al. (2007). Most of the motif-based T cell epitope prediction methods are straightforward mathematical analyses of the patterns of amino acids that occur in peptides bound to an HLA DQ allele by antigen-presenting cells. However, a notable limitation shared by all approaches is the possibility of producing false-positive and false-negative results.
Structural analyses involve the analysis of local secondary and tertiary structures, which help to identify structural epitopes as well as surface-positioned epitopes available for enzymatic digestability. Structural epitopes are built from short peptides distributed within a protein which result in allergic epitopes at the surface of the protein when it takes up its stable tertiary structure (Bredehorst and David 2001). Due to the nature of the HLA DQ–epitope interactions, structural analysis of T cell epitopes is mainly confined to the local structural characteristics of a linear epitope bound into the groove of the HLA DQ molecule. Based on our present knowledge, structural epitopes are not characteristic on celiac disease-related prolamins.
Structural B cell epitope prediction is an essential but unsolved problem in bioinformatics. Past surveys have estimated that only 10 % of the B cell epitopes are continuous (van Regenmortel 1996), but this ratio can be different for food and wheat allergens. Additionally, van Regenmortel (1996) noted that even linear epitopes adopt a conformational structure, and therefore, the distinction is somewhat blurred. Structural epitopes can be identified if the tertiary structure of a protein is available. Most of the prediction methods, such as BEpro (http://pepito.proteomics.ics.uci.edu/) and DiscoTope (http://tools.immuneepitope.org/stools/discotope/discotope.do) are using either known discontinuous epitope sequences or protein data bank (PDB) models of the allergens to identify or map structural epitopes.
To date, the amino acid distribution of the complementary wheat antigen surface has been difficult to characterize, which is partly due to the high number of distributed coiled regions present in wheat prolamins. This might also be related to the low number of prolamin superfamily members found in the PDB protein structure database. The relatively low number of allergen proteins with known tertiary structure cannot provide a sufficient base to develop high-throughput and accurate predictive systems. However, information gained from the secondary structures of the analyzed proteins may also help to understand sterical relations.
Structural domain composition, the number, and the distribution of alpha helical-, beta-turn structures, and coiled regions have an important role in the antigenicity predictions. The distribution of polar and apolar residues determines the hydrophilic domains which are likely to be exposed on the surface of a protein. Therefore, hydrophobicity plots can also be useful to identify potentially antigenic regions. Predicted structural domains for the prolamins clearly differentiate the sulfur-poor prolamins (high-molecular-weight (HMW) glutenins and omega gliadins) from sulfur-rich prolamins (alpha, gamma gliadins, and low-molecular-weight (LMW) glutenins). Sulfur-rich prolamins possess a relatively large C-terminal domain containing the conserved cysteine skeleton and a repetitive region in a similar length. The C-terminal domains of these proteins are rich in alpha-helices and form a compact, globular structure, stabilized by intraspecific disulfide bonds. In contrast, the repetitive region is usually formed from reverse beta-turns and polyproline II type helices. The overall structure of the repetitive domains highly depends on the consensus repeat motifs and the similarity between the different consensus sequences. In gamma gliadins, the repeat motifs are more conserved compared with the alpha gliadins. This results in a more extended repetitive domain for gamma gliadins when in solution (Shewry et al. 2009). In omega gliadins, where longer random coiled regions are interspersed with polyproline II helices, both extended and compact structures have been reported (Popineau and Pineau 1988; Shewry et al. 1997). HMW glutenins are also reported to have extended structures in solutions (for detailed review, see Shewry et al. 2009). The common characteristic of the repetitive regions present in the different cereal prolamins is that they are rich in residues like proline, glutamine, and phenylalanine. These residues are also the main contributors both in celiac and wheat allergy-related epitopes.
A significant consideration in predicting epitopes is whether or not a protein/peptide sequence in the database carries a proteolytic enzyme digestion site within the epitope that would destroy its efficacy as an epitope. The large proline content of prolamin proteins causes unusual secondary structure of the polypeptides that result in a poor digestibility and can confound the interpretation of the presence or absence of enzyme digestion sites. To avoid this limitation, the screening for the presence of epitopes in protein sequences is best carried out at both undigested and digested protein sequences derived from an in silico digestion. Such a twin screening procedure provides a maximum and minimum value of the number of epitopes in the protein database under study. There exists a large level similarity between the epitope structures and a large number show overlapping and embedded sequences in the prolamins (Salentijn et al. 2012). An alpha gliadin derived 33mer and some 29- and 26mers originated from gamma gliadins all contain some overlapping celiac disease-related epitopes (Shan et al. 2002; Salentijn et al. 2012) which were identified in an alpha gliadin. The HLA DQ alleles recognizing these epitopes belong to the same class (HLA DQ2); therefore, it is not clear whether these epitopes are competing or the actual structure of the peptide determines which of these epitopes will bind into the groove of a HLA DQ molecule. The adjacent residues might also have influence on T cell-stimulating capacity (Salentijn et al. 2012).
Prediction using a fully sequenced genome
The grass Brachypodium distachyon serves as the evolutionary closest sequenced genome to Triticeae (Vogel et al. 2010). Brachypodium possesses one of the smallest genomes of any grass and is suitable for both functional and structural genomic research. Brachypodium is also a suitable base to develop a workflow to identify and classify proteins that are responsible for wheat-related disorders such as celiac disease or wheat allergy (Juhász et al. 2012), due to its evolutionary relationships to wheat and barley. Unlike in Triticeae endosperm, globulin-like proteins are the main reservoir proteins in Brachypodium seeds. Classical prolamins express at lower levels than their homologs in wheat or barley consisting mainly of gliadin-like proteins, although some LMW and HMW glutenin-like proteins have also been identified (S. Wang, unpublished). Based on these characteristics, B. distachyon has some potential as a food source for patients suffering in gluten-related disorders. In the study of Juhász et al. (2012), linear, non-pollen epitopes originating from Pooideae species were retrieved from the Immunepitope database (www.immuneepitope.org). The obtained epitopes were mapped to the Brachypodium protein sequence database v1.2. Potential seed allergens were identified using developing Brachypodium seed ESTs and rice homologues. Trypsin, chymotrypsin, and pepsin were used in the in silico digestion experiment, and epitopes resistant to enzymatic cleavage were identified. Altogether, in 206 predicted proteins accounting for approximately 0.6 % of the entire Brachypodium proteome, at least one epitope causing either celiac disease or wheat allergy or both was found. Based on the epitope annotations, six epitopes were found to be related to celiac disease, and eight proteins could be assigned to wheat allergies. Molecular function of allergen proteins was evaluated using gene ontology terms. Among proteins containing CD epitopes, only about 6 % of the proteins fulfilled a seed storage function, and no storage proteins have been identified with WA epitopes. The majority of the proteins were involved in nucleic acid binding, transcription factors binding or activity, and protein dimerization.
Definition of expressed epitope level and toxicity strength using published proteomic data
The wheat proteome datasets published by Dupont et al. (2011) and Tasleem-Tahir et al. (2012) provide the necessary information to carry out an estimation of allergen prediction for a single cultivar. The workflow of the analyses carried out on these published data is presented in Fig. 1. Total protein extract originating from ripened Butte-86 seeds were used from the analysis of Dupont et al. (2011) while the water-soluble proteins were used from the analysis of cv. Recital (Tasleem-Tahir et al. 2012). Only proteins present in the mature seed were used.

Workflow of proteome-based allergen identification
Due to the level of similarity between the epitopes identified by different research groups, there exists a large number of epitopes which are overlapping or embedded sequences. There is a limited knowledge on the structural details of the cell/epitope interaction, and effects of these structural similarities are not known in detail. Because of this limitation in knowledge, an algorithm and executable software written in PowerBasic (Békés, unpublished) was used to ensure that any one amino acid in the protein/peptide sequence was counted only once during screening. Although this is a stringent approach to screening for T and B cell epitopes, it provides an estimate of the minimum number of epitopes in the proteins/peptide database.
In the light of the known molecular/immunological background of the biological response in case of CD, the level of toxicity has been related to the number of tTG sites in the epitope (Henderson et al. 2007). For example, the significantly larger effects of the 33mer found in an alpha gliadin compared with other epitopes can be explained by the presence of two tTG sites. Therefore, CD epitopes has been screened for the presence of tTG sites. Using the number of tTG sites as a weighting factor and counting the number of CD-epitopes per se found in a protein provided a number which could be used as a measure of the total level of toxicity in the protein. In case of WA epitopes, there was insufficient background knowledge to apply this metric.
Screening for the number of epitopes in the amino acid sequence of a protein/peptide provides the number of molar equivalent epitopes per molar equivalent of protein. In order to define the toxic/allergic level of a protein/peptide, both the “strength” of the individual epitopes and the relative expression levels of the proteins in the sample need to be considered.
The expression levels of the different proteins have been calculated from the spot volume data published by Dupont et al. (2011). We note that the use of calculated spot values is based on Coomassie Brilliant Blue staining. Although the number of bound dye molecules is approximately proportional to the protein amount, the dye molecules favor basic amino acids which can result slight differences in staining of proteins (Rigetti and Chillemi 1978; Tal et al. 1980). The amino acid composition of prolamins shows significantly less lysine and arginine residues than proteins usually. This reduced content of basic amino acids can result in differences in the amount of bound Coomassie Brilliant Blue molecules. Since 80 % of the seed endosperm proteins consist of prolamins, a correction based on the number of basic amino acid in the total amino acid number can be applied with a corrected spot volume data calculated as follows:
Where C i is the relative mass of protein i in the sample; [Spot Area]p,i is the spot area of the protein i; f i is the correction factor adjusting staining intensity of a spot based on the basic amino acid content of the protein i and is calculated as follows:
\( n_i^{+} \) is the number of positively charged amino acids in protein i; \( n_i^T \) is the total number of amino acids in protein i.
Using these normalized spot volume values, the epitope content of a sample can be calculated based on the following formula either in molar (X) or in mass (Y) units:
Where X and Y are the epitope contents of a sample in micromoles per 100 g protein and parts per million (ppm) units, respectively; [Mw] p,i is the molecular mass of protein i ; [n] e,i,j is the number of j epitopes found in protein i; [Mw] e,j is the molecular mass of epitope j; n is the number of proteins in the sample; m is the number of epitopes scanned for in the proteins; w j is the weighting factor. The value of w j is 1 for WA epitopes while, in case of CD epitopes, it indicates the number of “core epitopes” in the epitope j.
The protein composition has been expressed in mole-percent units, using the molecular mass of each identified spot group by taking the total amount of adjusted spot values on the gel as 100 %. These mole-percent data have been multiplied with the number of epitopes found during the screening process and finally converted to micromoles epitope per 100 g protein units. These normalized values establish the basis for comparisons among proteins and protein subgroups. The sum of these normalized values for a sample estimated the overall toxicity of the sample. The “epitope toxicity” value obtained in this way is a significant research output from the analysis of large datasets that can be applied to the food industry.
The epitope sources
The epitopes from the IEDB
Epitope selection targeted linear epitopes because these are unambiguous to assay compared with compound epitopes that required a 3D- or predicted 3D-structure. All epitopes used in the analyses are presented in the Electronic supplementary material, Table 1.
Immune Epitope Database (IEDB) (http://immuneepitope.org) was used to retrieve epitopes identified from Pooideae, similarly as presented in Juhász et al. (2012). Pollen-related epitopes were excluded from the analysis.
The antibody response as specified in the annotation of the epitope was used to differentiate CD and WA epitopes and yielded 549 CD epitopes and 174 WA epitopes. As mentioned above, the mapping of epitopes to identified protein/peptide sequences was carried out using the epitope conservancy analysis tool at IEDB tool center. The results were combined with the in-house-developed algorithm to exclude overlapping epitopes. The Butte86 and Recital databases contained 105 and 42 wheat protein sequences with at least one epitope in their sequences. Using the in silico enzymatic digestion process variation reduced the CD and WA epitopes being considered to 99 and 47, respectively. The number of tTG sites was also determined for each of the non-overlapping epitopes.
The 549 IEDB CD epitopes were separated into three subgroups based on whether they contained tTG binding sites and/or any of the “core CD epitopes” (for details, see below). The three subgroups were determined as follows:
-
1.
−tTG/−core: targets without both the tTG sites and core epitopes.
-
2.
+tTG/−core: A number of tTG sites were found that were not embedded into the core epitopes.
-
3.
+tTG/+core: targets containing both tTG sites and core epitopes.
The −tTG/−core group was the most frequent CD epitope subgroup, followed by the +tTG/+core group.
The Butte 86 proteins showed 617 CD epitopes and 435 wheat allergy-related epitopes. The number of CD epitopes decreased by almost 47 % when digested proteins were analyzed. The distribution of CD subsets was as follows: The 334 epitopes were found without core sites and tTG targets; 60 epitopes contained at least one tTG target amino acid but no core epitopes; 223 epitopes contained both tissue tTG targets and core epitope sites. The epitope numbers in the second and third subgroups were particularly sensitive to proteolytic digestion and decreased to approximately one third of the original epitope list after digestion. Proteins identified from cv. Recital did not contain any celiac disease-related epitopes.
In the Recital study, WA-related protein set 46 % of the epitopes was lost when digested proteins were examined. The number of WA-related epitopes was only 12 in the undigested set, and the number of detected epitopes decreased to seven when proteins were digested.
The core epitope set
The collection of CD core epitopes was based on Sollid et al. (2012) and using the 29 nonapeptides defined in the study, each nine-amino-acid in length. Similar to the IEDB set epitope analyses, the mapping was carried out both on undigested and digested proteins. No CD core epitopes could be mapped to the proteins in Recital. The number of core epitope hits was 109 in the undigested Butte 86 proteins. This count decreased to the half when mapping was carried out on digested polypeptides.
Predicted epitope list and T cell epitope prediction using MHC II binding predictions on IEDB
T cell epitope prediction has been carried out both on Butte86 and Recital protein sequences using the MHC II binding tool at IEDB Analysis Resource. HLA alleles DQ2.5 and DQ8 were used for the analysis. The number of predicted epitopes was much lower than expected for both protein sets. In Butte 86, altogether 36 hits were counted from the undigested set, and the number of detected predicted T cell epitopes was only four for the digested protein set. The respective number for the Recital proteins was 25 in the undigested protein set, and only two predicted epitopes were identified from digested proteins. Distribution of predicted epitopes among the different proteins is presented in the Electronic supplementary material, Table 3.
Epitope toxicity and epitope strength analyses
Epitope number and protein expression level relations have been evaluated at three levels:
-
1.
Most toxic proteins were described as a function of proteins with the largest epitope number in their sequence multiplied with their expression level. To highlight the importance of expression level on toxicity, proteins from Butte 86 have been graded based on the number of epitopes detected in their sequences, followed by an analysis of the epitope numbers multiplied by the expression levels.
-
2.
Most frequent CD, CD core, and WA epitopes have been identified from Butte 86 proteome.
-
3.
Epitope toxic levels were described as a function of the sum of individual epitopes in the entire proteome combined with the total expression levels.
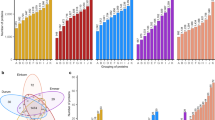
Most of the analyses were carried out on both proteomes using all the possible epitope lists. Proteins of the Butte 86 set were evaluated both at the individual level as well as in groups of related proteins. Proteins belonging to the same protein family were numbered continuously (e.g., LMW glutenin 1 to LMW glutenin 11). Details for relating protein IDs the original spot numbers are presented in Electronic supplementary material, Table 2. Among the 105 proteins analyzed, only the members of the main prolamins (HMW and LMW glutenins, alpha, gamma, and omega gliadins) and some proteins, which also belong to the prolamin superfamily, contained at least one celiac disease- or wheat allergy-related epitope. These proteins belong to alpha amylase inhibitors, lipid transfer proteins, and farinins (Fig. 2). Among the prolamin family members, the omega and gamma gliadins were the most dominant in CD epitope scores, with 20–25 epitopes in their sequence. If requirements of tTG target sites and the presence of nine-amino-acid-long core epitopes were also considered, omega gliadins (Omega gliadin 4 and Omega gliadin 5) and HMW GS Dx5 possessed the most +tTG/+core type CD epitopes. LMW glutenins possessed 5–16 CD epitopes; however, most of these epitopes did not contain tTG target amino acids or core epitopes. The undigested epitope numbers were decreased by about 50 % after digestion in most of the proteins. In HMW glutenins and several omega gliadins, all of the CD epitopes were digested by one of the enzymes. When digested profiles were considered, the Omega gliadin 2 and some of the LMW glutenins possessed the largest number of CD epitopes. Wheat allergy-related proteins were found among HMW glutenins, omega gliadins, gamma gliadins, and some of the LMW glutenins. Additionally, WA epitopes were identified in several alpha amylase inhibitors (WMAI and WDAI 1, 2, 3) and also in a lipid transfer protein. Enzymatic digestion did not affect the number of toxic epitopes as dramatically as in the CD-related proteins. When the total sum of CD and WA epitopes are considered, the HMW glutenins and some of the omega gliadins possess the most harmful polypeptides in their sequence.

Epitope counts in undigested and digested Butte 86 protein sets. WA epitopes are labeled in blue. Subgroups of CD epitopes, with the absence or presence of tTG sites and core epitopes are labeled in dark red (−tTG/−core), orange (−tTG/+core), and red (+tTG/+core). Undigested protein sets were used in the upper panel, and digested proteins are presented in the lower panel
The dataset of water-extractable proteins of cv. Recital showed some overlap with the Butte 86 set. The protein set contained many alpha amylase inhibitors, several serpins, heat-shock proteins, and a number of enzymes, such as beta amylases, protein disulphide isomerases, chitinases, and glutamate dehydrogenase. For detailed list of proteins, see the Electronic supplementary material, Table 2. None of these proteins contained celiac disease epitopes, and only six wheat allergy-related proteins could be identified. All of these proteins belong to the alpha amylase inhibitors. Four proteins are members of the dimeric alpha amylase inhibitors, and two of them belong to monomeric alpha-amylase inhibitors. The number of identified WA epitopes was two in each protein, and the number of intact epitopes after digestion was one for all protein. Although only alpha-amylase inhibitors contained WA epitopes from the Recital, there is a number of further non-prolamin allergens reported from wheat (Tatham and Shewry 2008; Salcedo et al. 2011; Altenbach et al. 2007). The absence of further allergens might be due to differences in solubility or genetic effects. Therefore, further analyses targeting the epitope toxicity and epitope strength analyses have been carried out on the larger Butte 86 dataset.
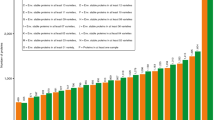
When most toxic proteins were compared using not only the epitope counts but also the expressed amounts of proteins, the relative order of toxic proteins changed significantly (Fig. 3). The expressed amount of epitopes in millimoles was normalized to 100 g proteins. The relative toxic level of HMW glutenins was much lower when only epitope counts were considered. This fact was due to the low expression level of HMW glutenins compared with LMW glutenins or the sulfur-rich alpha and gamma gliadins. In contrast, the toxic effect of some of the LMW glutenins was significant, especially when celiac disease-related epitopes were considered. The LMW glutenin 4, which was identified as an m-type LMW glutenin (LMW glutenin Bu-8 in Dupont et al. (2011)) is the most toxic protein, followed by several omega gliadins and further LMW glutenins. Among the HMW glutenins, the subunit Bx7 is the most toxic. When only wheat allergy-related proteins were analyzed, one of the omega gliadins (Omega gliadin 1) was the most toxic. The sequence of this omega gliadin is identical with the omega5 gliadin that is known as the most toxic protein for inducing WDEIA. The HMW glutenins are also highly toxic in terms of wheat allergy. For both the omega gliadin and the HMW glutenins, no significant loss of toxicity was observed when digested. In contrast, the toxic effect of LMW glutenins related to WA was only significant if undigested proteins were considered. The toxic level of gamma and alpha gliadins has slightly lowered when digested, although these proteins are not considered significant contributors to WA.

Epitope expression levels of CD and WA epitopes in digested (lower panel) and undigested (upper panel) Butte 86 protein sets. CD epitopes are labeled in red; WA epitopes are labeled in blue. Epitope expression levels are calculated in millimoles epitope per 100 mg protein values
Most frequent epitopes were evaluated on two epitope sets using the Butte 86 proteins. The results obtained from the IEDB celiac disease and wheat allergy assigned epitope sets are summarized in the Table 1. The significance of calculating the total expression levels of individual epitopes was particularly evident from the analysis of most toxic WA epitopes in the Butte 86 proteome. Ten epitopes with the highest number of counts in the entire Butte 86 set were evaluated at the expression level. Amounts of epitopes were calculated both in undigested and digested proteins. Expression levels for individual epitopes were summarized in each prolamin family (Fig. 4). The order of epitopes in Fig. 4 represents the order of epitope counts (Table 1). WA epitopes with the three highest frequencies in the undigested proteins were QQPFP, QQPGQ, and QQQPP. Based on their expressed amounts, epitope QQQPP seems to be more toxic compared with QQPGQ in the undigested proteins. When proteins were digested, the toxic epitope QQPGQ became harmless. The most toxic epitope was QQPFP, which was present in all the major prolamins except for HMW glutenins. The epitope QQQPP is characteristic in LMW glutenins, and based on their expression level, QQQPP is the second most toxic epitope. When epitope amounts are expressed in grams using their calculated molecular weight, the expressed amounts of toxic polypeptides is comparable to the ppm values used in the food industry to determine gluten-free products. The three most toxic WA epitopes expressed in micrograms per 100 g proteins represents 2.91, 1.25, and 2.22 ppm toxicity. This interpretation of the toxicity level of a sample is incomparably more exact and precise than the present food industry practice, critically reviewed by Wieser (2008). Analysis of 29 core celiac disease epitopes, all nine-amino-acid in length and containing at least one tTG target site, has been also carried out in order to relate amounts of epitopes directly to unique HLA DQ alleles. HLA DQ assigned epitopes were detected in “classic” prolamins, and no epitopes were detected in minor prolamins. This might be due to the fact that minor prolamins like farinins and purinins contain the conserved cysteine skeleton domains in a reduced length when compared with the glutenins. Gamma gliadins contain the largest number of DQ2.5-related epitopes. HLA DQ2.5 epitopes are present in LMW glutenins, gamma, omega, and alpha gliadins. The DQ8-related epitopes are also present in a significant number in HMW glutenins, and some of them have also been detected in alpha gliadins.

Expression levels for individual epitopes in each prolamin family. Expression levels of the ten most harmful wheat allergy-related epitopes are presented in undigested and digested Butte 86 protein sets. Expression levels are calculated in millimoles per 100 mg protein. Frequency of the epitopes in the different prolamin families is labeled by different colors
Two from the most frequent core epitopes were assigned to HLA DQ2.5 alleles. PQPQQQFPQ has been identified in 31 cases in the 105 proteins, while PQPQQPFPW was identified 12 times in the 105 proteins. The PQPQQQFPQ epitope was the most frequent in gamma gliadins and omega gliadins while PQPQQPFPW epitopes were the sole characteristic of LMW glutenins. The most frequent HLA DQ8 epitope, QGYYPTSPQ, has been found in 21 cases (Fig. 5) and was only characteristic of HMW glutenins. When expression levels were also considered, the toxicity of HLA DQ8-related proteins decreased, because of the low expression levels of HMW glutenins compared with sulfur-rich prolamins, which contained more HLA DQ2.5-associated epitopes. HLA DQ2.5-associated epitopes were mainly found in gamma gliadins. Sequences of different gamma gliadins contained about four times the amount of epitopes found in LMW glutenins or alpha and omega gliadins. HLA DQ8-assigned epitopes were characteristic on HMW glutenins, but some epitopes have also been detected in alpha gliadins.

Frequency and expression levels of core CD epitopes in the Butte 86 protein set. Epitopes are grouped based on their affinity to HLA DQ2.5 and DQ8 alleles
When proteins were digested, the number of intact epitopes in all of the above-mentioned cases dramatically decreased, and the HLA DQ8-allele-related HMW glutenin epitopes, in particular, disappeared. The most toxic epitope that remained was the HLA DQ2.5 PQPQQQFPQ epitope with 24 occurrences, followed by HLA DQ2.5 PQPQQPFPW (seven occurrences), and HLA DQ8 QGYYPTSPQ (five occurrences), respectively. Due to the smaller intact epitope number and the expression levels, the toxic levels of the most harmful epitopes were about 77 % of the undigested protein set.
Structural analysis of toxic and allergen proteins in Butte 86
When the secondary structure elements of the most toxic proteins and distribution of CD and WA epitopes were compared, the presence of epitopes was significant in the repetitive regions. Sulfur-rich prolamins like alpha, gamma gliadins, and LMW glutenins possessed several CD and WA epitopes in their C-terminal domains; however, these epitopes were mostly located in the coiled regions of the C-terminal domain. Sulfur-poor prolamins, like omega gliadins and HMW glutenins, possess a large repetitive region with high frequency of coiled regions. The distribution of both CD and WA epitopes was scattered throughout the entire sequence. An example for a sulfur-rich prolamin (alpha gliadin) and an omega gliadin is presented in Fig. 6.

Secondary structure elements and position of identified epitopes in prolamins. Both secondary structure predictions and epitope mappings have been carried out using CLC Genomic Workbench. Gray shadows in secondary structures represent alpha helical domains; dark grey blocks represent beta turn motifs. CD mapped CD epitopes, labeled in red; CD_dig mapped epitopes in digested proteins are labeled in orange; WA wheat allergy epitopes are labeled with blue blocks; WA_dig WA epitopes intact after digestion are labeled in light blue
Most of the epitopes were located in the repetitive regions. The length of this repetitive region is variable in the different prolamin families but results in similar structures with reverse beta turns and polyprolil helices dispersed in random coiled regions. There were also some epitopes present in the C-terminal domains of sulfur-rich prolamins. Those which are stable under digestive conditions are located in the random coiled inserts of the C-terminal. There are some WA epitopes which are located in alpha helix regions, but most of them can be digested with one of the endopeptidases.
Utilizing wheat proteome data can have a significant role when single cultivars are evaluated for wheat allergy or toxicity resulting in celiac disease. The epitope analyses of gel-based or MS-based data requires precise protein identification and a reliable determination of expression values. When members of the prolamin protein family are analyzed, the high homology observed in the single prolamin families also confirms the need for precise protein identification. Next to the analysis of single cultivars, there is a high potential in the analysis of whole grain, wheat flour blends, or end-products from different environments. Introducing the terms epitope expression levels or toxic strength may provide a more correct determination of standards for gluten-free products.
References
Akagawa M, Takeshi M, Shigenori I, Morita N, Suyamato K (2007) Proteomic analysis of wheat flour allergens. J Agric Food Chem 55:6863–6870
Altenbach SB, Kothari KM, Tanaka CK, Hurkman WJ (2007) Genes encoding the PR-4 protein wheatwin are developmentally regulated in wheat grains and respond to high temperatures during grainfill. Plant Sci 173:135–143
Amano M, Ogawa H, Kojima K, Kamidaira T, Suetsugu S, Yoshihama M, Satoh T, Samejima T, Matsumoto I (1998) Identification of the major allergens in wheat flour responsible for baker's asthma. Biochem J 330:1229–1234
Anderson RP, Wieser H (2006) Medical applications of gluten—composition knowledge. In: Wrigley CW, Békés F, Bushuk W (eds) Gliadin and glutenin. The unique balance of wheat quality. AACCI Press, St Paul, pp 387–409
Anderson RP, Degano P, Godkin AJ, Jewell DP, Hill AVS (2000) In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptid as the dominant A gliadin T-cell epitope. Nature (Med) 6:337–342
Armentia A, Martin-Santos JM, Blanco M (1990) Exercise induced anaphylaxis reaction to grain flows. Ann Allergy 65:149–151
Bredehorst R, David K (2001) What establishes a protein as an allergen? J Chromatogr B Biomed Sci Appl 756:33–40
Breiteneder H (2008) Can any protein become an allergen? Rev Fr Allergol Immunol Clin 2008(48):135–138
Breiteneder H, Radauer C (2004) A classification of plant food allergens. J Allergy Clin Immunol 113:821–830
Brusic V, Rudy G, Honeyman G, Hammer J, Harrison LC (1998) Prediction of MHC class II-binding peptides using an evolutionary algorithm and artificial neural network. Bioinformatics 14:121–130
Brusic V, Petrovsky N, Zhang G, Bajic VB (2002) Prediction of promiscuous peptides that bind HLA class I molecules. Immunol Cell Biol 80:280–285
Brusic V, Petrovsky N, Gendel SM, Millot M, Gigonzac O, Stelman SJ (2003) Computational tools for the study of allergens. Allergy 58:1083–1092
Brusic V, Bajic VB, Petrovsky N (2004) Computational methods for prediction of T-cell epitopes—a framework for modelling, testing, and applications. Methods 34(4):436–443
Catassi C, Fasano A (2008) Celiac disease. In: Arendt EK, Dal Bello F (eds) Gluten-free cereal products and beverages. Academic Press, San Diego, pp 1–28
Chou PY, Fasman GD (2006) Prediction of the secondary structure of proteins from their amino acid sequence. In: Meister A (ed) Advances in enzymology and related areas of molecular biology, vol 47. John Wiley & Sons, Inc, Hoboken
Davies IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383, 10.1093/nar/gkm216
Donnes P, Elofsson A (2002) Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinformatics 3:25
Doytchinova IA, Walshe V, Borrow P, Flower DR (2005) Towards the chemometric dissection of peptide-HLAA* 0201 binding affinity: comparison of local and global QSAR models. J Comput Aided Mol Des 19:203–212
Dupont FM, Vensel W, Tanaka CK, Hurkman WJ, Altenbach SB (2011) Deciphering the complexities of the wheat flour proteome using quantitative two-dimensional electrophoresis, three proteases and tandem mass spectrometry. Proteome Sci 9:10
Emini EA, Hughes JV, Perlow DS, Boger J (1985) Induction of hepatitis A virus neutralizing antibody by a virus-specific synthetic peptide. J Virol 55:836–839
Garsed K, Scott BB (2007) Can oats be taken in a gluten-free diet? A systematic review. Scand J Gastroenterol 42(2):171–178
Green PH, Cellier C (2007) Celiac disease. N Engl J Med 357:1731–1743. doi:10.1056/NEJMra071600
Henderson KN, Tye-Din JA, Reid HH, Chen Z, Borg NA, Beissbarth T, Tatham A, Mannering SI, Purcell AW, Dudek NL, van Heel DA, McCluskey J, Rossjohn J, Anderson RP (2007) A structural and immunological basis for the role of human leukocyte antigen DQ8 in celiac disease. Immunity 27:23–34
Juhász A, Gy G, Sebestyén E, Haraszi R, Tamás L, Balázs E (2012) Brachypodium distachyon as a model for defining the allergen potential of non-prolamin proteins. Funct Integr Genomics 12:439–446. doi:10.1007/s10142-012-0294-z
Karplus PA, Schultz GE (1985) Prediction of chain flexibility in proteins. Naturwissenschaften 72:212–222
Kolaskar AS, Tongaonkar PC (1990) A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 276(1–2):172–174
Laurièrre M, Pecquet C, Bouchez-Mahiout I, Snegaroff J, Bayrou O, Raison-Peyron N, Vigan M (2006) Hydrolyzed wheat proteins present in cosmetics can induce immediate hypersensitivities. Contact Dermatitis 54:283–289
MacNamara A, Kadolsky U, Bangham CRM, Asquith B (2009) T-cell epitope prediction: rescaling can mask biological variation between MHC molecules. PLoS Comput Biol 5(3):e1000327
Mills ENC, Madsen C, Shewry PR, Wichers HJ (2003) Food allergens of plant origin—their molecular and evolutionary relationships. Trends Food Sci Technol 14:145–156
Moolhuijzen P (2011) Bioinformatics approaches for functional predictions in diverse informatics environments. PhD Thesis. Murdoch University, Perth
Osorio C, Wen N, Gemini R, Zemetra R, Wettstein D, Rustgi S (2012) Targeted modification of wheat grain protein to reduce the content of celiac causing epitopes. Funct Interg Genomics 12(3):417–438. doi:10.1007/s10142-012-0287-y
Parker JM, Guo D, Hodges RS (1986) New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 25:5425–5432
Popineau Y, Pineau F (1988) Changes of conformation and surface hydrophobicity of gliadins. Lebensm Wiss Technol 21:113–117
Radauer C, Breiteneder H (2007) Evolutionary biology of plant food allergens. J Allergy Clin Immun 120:518–525
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanović S (1999) SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213–219
Rigetti AC, Chillemi GF (1978) Isoelectric focusing of peptides. J Chrom 156:243–251
Salcedo G, Quirce S, Diaz-Perales A (2011) Wheat allergens associated with baker’s asthma. J Investig Allergol Clin Immunol 21(2):81–92
Salentijn EMJ, Mitea DC, Goryunova SV, van der Meer IM, Padioleau I, Gilissen LJWJ, Koning F, Smulders MJM (2012) Celiac disease T-cell epitopes from gamma-gliadins: immunoreactivity depends on the genome of origin, transcript frequency, and flanking protein variation. BMC Genomics 13:277
Sampson HA, Metcalfe DD (1992) Food allergies. J Am Med Assoc 268:2840–2844
Sapone A, Bai JC, Ciacci C, Dolinsek J, Green PHR, Hadjivassiliou M, Kaukinen K, Rostami K, Sanders DS, Schumann M, Ullrich R, Villalta D, Volta U, Catassi C, Fasano A (2012) Spectrum of gluten-related disorders: consensus on new nomenclature and classification. BMC Med 10:13. doi:10.1186/1741-7015-10-13
Shan L, Molberg Ø, Parrot I, Hausch F, Filiz F, Gray GM, Sollid LM, Khosla C (2002) Structural basis for gluten intolerance in celiac sprue. Science 297(5590):2275–2279
Shewry PR, Miles MJ, Thomson NH, Tatham S (1997) Scanning probe microscopes—applications in cereal science. Cereal Chem 74:193–199
Shewry PR, D’Ovidio R, Lafiandra D, Jenkins JA, Mills NF, Békés F (2009) Wheat grain proteins. In: Khan K, Shewry PR (eds) Wheat chemistry and technology. AACC Press, St Paul, pp 223–298
Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F (2012) Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics 64:455–460. doi:10.1007/s00251-012-0599-z
Tal M, Silberstein A, Nusser E (1980) Why does Coomassie Brilliant Blue R interact differently with different proteins? A partial answer. Biol Chem 260:9976–9980
Tasleem-Tahir A, Nadaud I, Chambon C, Branlard G (2012) Expression profiling of starchy endosperm metabolic proteins at 21 stages of wheat grain development. J Proteome Res 11:2754–2773
Tatham AS, Shewry PR (2008) Allergens in wheat and related cereals. Clin Exp Allergy 38:1712–1726
Tomar N, De RK (2010) Immunoinformatics: an integrated scenario. Immunology 131:153–168
Van Heel DA, West J (2006) Recent advances in coeliac disease. Gut 55:1037–1046
Van Regenmortel MHV (1996) Mapping epitope structure and activity: from one-dimensional prediction to four-dimensional description of antigenic specificity. Methods 9:465–472
Vogel JP et al (2010) Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463:763–768
Wieser H (2008) Detection of gluten. In: Arendt EK, Dal Bello F (eds) Gluten-free cereal products and beverages. Academic Press, San Diego, pp 47–80
Acknowledgments
The authors wish to thank Professor Rudi Appels for the motivating discussions and suggestions during the analyses and the preparation of the manuscript. The Project is supported by the European Union and co-financed by the European Social Fund (grant agreement no. TÁMOP-4.2.2.A-11/1/KONV-2012-0008).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Epitope lists used in the analyses are presented as IEDB_CD epitopes, IEDB_WA epitopes; core epitopes based on Sollid et al. 2012; predicted T cell epitopes using both Butte86 and Recital protein sets (XLSX 49 kb)
Supplementary Table 3
Distribution of different epitope sets in Butte 86 and Recital databases. CD epitope subtypes containing tTG sites and core epitopes are also represented in separate columns, both for digested and undigested protein sets (XLSX 18 kb)
Rights and permissions
About this article
Cite this article
Juhász, A., Gell, G., Békés, F. et al. The epitopes in wheat proteins for defining toxic units relevant to human health. Funct Integr Genomics 12, 585–598 (2012). https://doi.org/10.1007/s10142-012-0302-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-012-0302-3