
Abstract
Maize (Zea mays ssp. mays L.) is an important model organism for fundamental research in the agro-biotechnology field. Aldehydes were generated in response to a suite of environmental stresses that perturb metabolism including salinity, dehydration, desiccation, and cold and heat shock. Many biologically important aldehydes are metabolized by the superfamily of NAD(P)+-dependent aldehyde dehydrogenases. Here, starting from the database of Z. mays, we identified 28 aldehyde dehydrogenase (ALDH) genes and 48 transcripts by the in silico cloning method using the ALDH-conserved domain amino acid sequence of Arabidopsis and rice as a probe. Phylogenetic analysis shows that all 28 members of the ALDH gene families were classified to ten distinct subfamilies. Microarray data and quantitative real-time PCR analysis reveal that ZmALDH9, ZmALDH13, and ZmALDH17 genes involve the function of drought stress, acid tolerance, and pathogens infection. These results suggested that these three ZmALDH genes might be potentially useful in maize genetic improvement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aldehydes were generated in response to a suite of environmental stresses that perturb metabolism including salinity, dehydration, desiccation, and cold and heat shock. The aldehyde dehydrogenase (ALDH) superfamily comprises a group of divergently related enzymes that metabolize a wide spectrum of endogenous and exogenous aldehydes (Sophos and Vasiliou 2003). Many aldehydes are chemically reactive and toxic at physiological concentrations. However, active ALDH enzymes can detoxify the aldehydes by oxidation to their corresponding carboxylic acids (Kirch et al. 2004). In addition, it has been reported that some ALDH proteins were identified as binding proteins which were capable of non-catalytic interactions with chemically diverse endogenous or exogenous compounds (Sophos and Vasiliou 2003). The ALDH Gene Nomenclature Committee (AGNC) has established the criteria for cataloging deduced ALDH protein sequences (Vasiliou et al. 1999). Based on these criteria, protein sequences with more than 40 % identity to a previously identified ALDH sequence represent a family, and sequences with more than 60 % identity compose a protein subfamily. Protein sequences that have less than 40 % identity would describe a new ALDH protein family (Kirch et al. 2004). ALDHs are found in both prokaryotes and eukaryotes. The eukaryotic ALDHs can be organized into 21 families based on sequence identity (Sophos and Vasiliou 2003). In mammals, different ALDH representatives have been implicated in intermediate metabolism, such as vitamin A biosynthesis and amino acid metabolism, as well as in detoxification of stress-generated aldehydes and osmoprotection (Perozich et al. 1999). The plant ALDH genes are represented in 12 ALDH families: ALDH2, ALDH3, ALDH5, ALDH6, ALDH7, ALDH10, ALDH11, ALDH12, ALDH18, ALDH19, and ALDH21. The ALDH11, ALDH19, ALDH22, and ALDH21 families are unique to plants (Kirch et al. 2004; Rodrigues et al. 2006; Kotchoni et al. 2012). Biological function has been assigned to some members of the plant ALDH superfamily in development and stress adaptation (Rodrigues et al. 2006). Among the stress-related ALDHs, the plant ALDH3 and ALDH5 families are involved in detoxification of aldehydes (Bouché et al. 2003; Sunkar et al. 2003), whereas the ALDH10, ALDH11, and ALDH12 families act primarily in cellular osmotic regulation by catalyzing the synthesis of osmoprotectant (Kirch et al. 2004). With the genome of more organisms being fully sequenced, the numbers of ALDH genes identified have increased lately. We are interested in understanding the molecular and genetic bases of drought stress and disease resistance in maize with a particular emphasis on the aldehyde dehydrogenase (ALDH) protein superfamily. In maize, only a few ALDH genes have been characterized, and the functions of most of them remain to be determined. Completion of the high-quality sequencing of the maize genome has provided an excellent opportunity for genome-wide analysis of genes belonging to specific gene families. Here, we identified 28 ZmALDH genes in maize by database searches and classified these genes as belonging to ten subfamilies. We also performed a complete survey of genomic organization, chromosomal location, and sequence homology of all ZmALDHs; investigated differential gene expression patterns under various stress conditions; and investigated the phylogenetic relationship between ALDH genes in Arabidopsis and maize.
Materials and methods
Searching for ALDH genes and locations on chromosomes
Protein sequences of six known plant ALDHs (OsALDH18, AK101230; SrALDH21A1, AAK59374; ALDH10A8 NA, AY093071; ALDH7B4, AJ584645; ALDH5F1, AF117335) were used as queries to search against the protein database of maize in Genetics and Genomics (MaizeGDB, http://www.maizegdb.org/), Maize Sequence Search (http: //maizesequence. org/index.html), TIGR Maize Database (http://maize.jcvi.org/) and National Centre for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/). All sequences with an E value < 1e−6 were selected for manual inspection. Two ALDH active site signature sequences, glutamic acid active site (PROSITE PS00687) and cysteine active site (PROSITE PS00070), were also considered in this search (Kirch et al. 2004). The ALDH Pfam (Aldehyde_DH_dom, PF00171) search (http://pfam.sanger.ac.uk/, http://www.maizegdb.org/) was employed to confirm the candidate sequences as ALDH proteins. Exon and intron structures of this gene family were investigated using Maize Sequence Search (http://maizesequence.org/index.html) database. ZmALDH genes were located on maize chromosomes according to the positions specified in the database of Maize Sequence Search (http: //maizesequence.org/index.html).
Sequence alignments and phylogenetic analyses
Multiple protein alignment was performed with ClustalX 1.83 (Thompson et al. 1997). Alignment was edited manually using GeneDoc (Nicholas et al. 1997). Identification of conserved motifs of maize ALDHs was accomplished with multiple sequence alignments and Multiple Em for Motif Elicitation version 3.5.7 (http://meme.sdsc.edu). Phylogenetic and molecular evolutionary analyses were conducted using MEGA version 5, the parameters were set to multiple alignment gap opening penalty at 10, gap extension penalty at 0.2, and delay divergent cutoff at 30 % (Tamura et al. 2011). To generate a phylogenetic tree, complete ALDH-predicted proteins in Z. mays were aligned using the ClustalX version 1.83 (Thompson et al. 1997). The neighbor-joining method was used to construct different trees; pairwise deletion opinion and the Jones, Taylor, and Thornton (JTT) model for amino acid sequences were used (Saitou and Nei 1987). The reliability of the obtained trees was tested using bootstrapping with 500 replicates.
Microarray data collection and analysis of expression profile
The expression behaviors of maize ALDH genes were examined in a set of maize microarray data downloaded from GEO at NCBI (http://www.ncbi.nlm.nih.gov/geo) and the transcriptome data at PLEXdb (http://www.plexdb.org). The microarray data of maize gene expression during infection with Ustilago maydis (GSE10023) was generated by hybridization of RNAs from the samples of infected leaves with the solopathogenic U. maydis strain SG200 that were taken at 12 and 24 h post-infection, as well as 2, 4, and 8 days post-infection. Samples from uninfected control plants were taken at the same time points. The microarray data of genome-wide transcriptome analysis of two maize inbred lines under drought stress during the seedling stage (GSE16567) was generated from the transcriptome changes during drought tolerance in maize (drought-tolerant line Han21 and drought-sensitive line Ye478) at the seedling stage (Zheng et al. 2010). The microarray data and expression profile of contrasting maize genotypes grown on acid and control soil (root tips) (GSE21070) was collected from the transcriptional changes of two maize genotypes Cat100-6 (Al tolerant) and S1587-17 (Al sensitive) (Mattiello et al. 2010). For four genes (ZmALDH9, ZmALDH13, ZmALDH17, and ZmALDH18) with more than one unique probe, we selected the probe with higher intensity value for calculation.
Plant materials and quantitative real-time PCR
Maize (Zea mays L. inbred line B73) plants were grown at 28 ± 2 °C with a photoperiod of 16-h light and 8-h dark. Three-week-old seedlings (developed three fully opened trifoliate leaves) were used to investigate the effects of jasmonate (JA) treatments and salt stresses on annexin gene expression. For the JA treatments, seedlings were sprayed with solutions containing 50 μM JA (Sigma) dissolved in dimethylsulfoxide (DMSO). The control plants were mock-treated with DMSO. The seedling (including root, stem, and leaves) were collected after 0, 6, and 12 h. For the salt stress, the seedlings were sprayed with 50 mM NaCl, and the control plants were sprayed with water. The seedlings were collected after 0, 7, and 14 day. All samples were quickly frozen in liquid nitrogen and stored at −80 °C until use.
Total RNA was prepared using Trizol reagent (Takara), followed by DNase I treatment to remove any genomic DNA contamination. The first-strand cDNA was synthesized from 1 μg of total RNA using PrimeScript™ RT reagent Kit with gDNA Eraser for RT-qPCR. Quantitative RT-PCR was carried out using an CFX96 manager real-time PCR system. Table 1 contains a list of primers of investigated genes. Each reaction contained 1 μL cDNA, 1 μL mixed primers, 5 μL SYBR Green PCR Master Mix (Takara), and 3 μL nuclease-free water. Reaction mixtures were incubated for 2 min at 50 °C, 1 min at 95 °C, followed by 40 cycles of 5 s at 95 °C, 30 s at 50 °C, and 2 min at 72 °C (Zhou et al. 2010a, 2011, 2012). The gene encoding the actin was used as control. At least three replicates of each cDNA sample were performed for quantitative RT-PCR analysis. Real-time PCR data were analyzed using the 2−ΔΔCt method (Zhou et al. 2010a, 2011, 2012).
Statistical analysis
All data were examined using one-way analysis of variance (ANOVA) (General Linear Models on GLM procedure). The Student–Neuman–Keuls test was used to identify the means which differed if the ANOVA test indicated significance. A P value <0.05 was considered to be significant (Zhou et al. 2010a, 2011, 2012).
Results
Maize ALDH superfamily contains 28 members
In this study, we grouped the maize ALDH proteins that had more than 40 % sequence identity with known ALDHs into the same family (Kirch et al. 2004). Twenty-eight maize genes were identified to encode members of ten ALDH families (Table 2 and Electronic supplementary material (ESM) Fig. S1). Six families (families 2, 3, 10, 5, 7, and18) contained multiple members, and each of the other four families (families 6, 11, 12, and 22) was represented by a single gene. In order to ensure the accuracy of the sequences used in the following work, a database search was carried out to find the transcripts matching the candidate ALDH sequences. Six genes contained multiple transcripts, including ZmALDH27, which contains five transcripts. The chromosomal locations and directions of transcription of each ALDH gene was, therefore, determined and demonstrated (Table 2 and Fig. 1). The family of 28 ZmALDHs is distributed on all ten maize chromosomes; but only one family member each are located on chromosomes 1, 8, and 9. Three ZmALDHs are present on chromosome 6; four each on chromosomes 3 and 10; five each on chromosomes 2 and 4; and two each on chromosomes 5 and 7. It is noteworthy that the nomenclature system for ZmALDHs used in the present study, a generic name from ZmALDH1 to ZmALDH28, was provisionally given to distinguish each of the ALDH genes according to its position from the top to the bottom on the maize chromosomes 1 to 10, which is a different nomenclature system than the one previously used (Wang et al. 2007). For ZmALDH27, which contains five transcripts, a genetic name from ZmALDH27.1 to ZmALDH27.5 was given to distinguish each of the ZmALDH27 transcripts. ZmALDH3 contains two transcripts ZmALDH3.1 and ZmALDH3.2; however, only ZmALDH3.2 has a Pfam (Aldehyde_DH_dom, PF00171) domain. The nomenclature system for ZmALDHs genes and transcripts used in this study could be easily accepted by researchers.

Genomic structures and phylogenetic relationship of 28 maize genes and 48 transcripts. The phylogenetic trees were constructed using MEGA 5.0. Only coding exons, represented by black boxes, were drawn to scale. Line angles connecting two boxes represent introns
Phylogenetic analysis of ALDH proteins
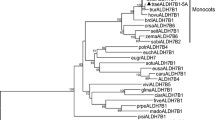
From the unrooted phylogenetic tree generated from alignments of the full-length protein sequences of all the ZmALDHs including different kinds of transcripts, it was found that all of the ZmALDHs fell broadly into ten major families with well-supported bootstrap values (Figs. 1 and 2). To examine the phylogenetic relationships of maize, Tortula ruralis, rice, and Arabidopsis ZmALDH proteins, a phylogenetic tree was constructed from alignments of the full-length protein sequences of 48 ZmALDHs, 1 TrALDHs, 4 OsALDHs, and 14 ALDHs. All 67 members of the ALDH gene families were classified into 11 major groups, representing the 11 distinct ALDH families (Fig. 2). Similar to the results of previous work (Kirch et al. 2004), families 2, 5, and 10 clustered together. Families 22 and 3 were connected by a node with a high bootstrap value, indicating a close relationship between them.

Phylogenetic relationships of maize, rice, and Arabidopsis ALDH proteins. The unrooted NJ tree was generated with the MEGA 5.0 program. Bootstrap values from 500 replicates are indicated at each branch. The nine major groups representing 11 distinct ALDH families are indicated
Intron loss might accompany the recent evolution of maize ALDH genes
A comparison of the full-length cDNA sequences with the corresponding genomic DNA sequences was made to determine the numbers and positions of exons and introns of each individual ZmALDH gene. It was found that the coding sequences of all the ZmALDH genes are disrupted by introns except for ZmALDH2. Intron loss was observed for maize ALDH genes in families 2, 7, and 18. In comparison with other genes in family 2, ZmALDH24 had acquired three more exons in the 5′-end during evolution. For ZmALDH2, no exon was observed.
Maize ALDH genes were differentially expressed under different stress
Plant ALDH genes have been identified to play important roles in the adaptation of plants to various abiotic stresses. We therefore examined the responses of maize ALDH genes to drought, acid stresses, and pathogen infection in young leaf tissue through the collection and analysis of microarray data. Moderate and severe drought stress repress ZmALDH9 (Fig. 3a) and ZmALDH17 (Fig. 3b) gene expression. However, re-watering can rescue the expression levels of these two genes. ZmALDH13 represents a higher level of gene expression in the genotype of S1587-17 (Al sensitive) compared to Cat100-6 (Al tolerant) (Fig. 4). In addition, acidic soil can activate ZmALDH13 gene expression (Fig. 4), which demonstrates that ZmALDH13 is involved in the function of acid tolerance. U. maydis inoculation with maize leaves can induce ZmALDH9 and ZmALDH18 expression (Fig. 5). These microarray data revealed that ZmALDH9 plays an important role in drought stress and pathogen infection.

Expression profiles of maize ALDH genes in maize inbred lines (Han21: drought tolerant; Ye478: drought sensitive) under drought stress during the seedling stage. a, ZmALDH9 (Zm.10336.1.S1_at). b, ZmALDH17 (Zm.8118.1.A1_at). Error bars are the standard deviations of three replicates

Expression profiles of ZmALDH13 (Zm.2793.1.A1_at) gene in maize genotypes (Cat100-6: Al tolerant; S1587-17: Al sensitive) grown on acid and control soil. Error bars are the standard deviations of three replicates

Expression profiles of ALDH genes in maize during infection with U. maydis. a, ZmALDH9 (Zm.10336.1.S1_at). b, ZmALDH18 (Zm.10312.1.A1_at). Error bars are the standard deviations of three replicates. dpi: days post-inoculation
To confirm the results of microarray data, four ZmALDHs (ZmALDH9, ZmALDH13, ZmALDH17, and ZmALDH18) were chosen for expression analysis by real-time RT-PCR under JA and NaCl treatment (Fig. 6). JA treatment represses ZmALDH9 and ZmALDH13 gene expression (Fig. 6a). All these genes were induced by salt stresses, except for ZmALDH18, which showed decreased levels in its transcription level under this stress (Fig. 6b). ZmALDH17 was the only strongly induced ALDH gene by salt stress, its expression level increased more than 50 times compared with the mock, suggesting that it may be related to the salt stress tolerance of the maize.

Expression patterns of four ALDH genes in maize under JA and NaCl treatments. White and black boxes represent mock and 50 μM JA (a) or 50 mM NaCl treatments (b), respectively. The numbers 6, 12, and 7, 14, represent the time hour and day after treatments, respectively. *P value <0.05. Error bars are the standard deviations of three replicates
Discussion
In this study, the maize genome database contains 48 transcripts encoding members of ten ALDH gene families (Table 2 and ESM Fig. S1), which are also represented in other angiosperm plants including rice, poplar, and grape (Gao, and Han 2009). Previous reports showed that the maize genome contains 24 unique ALDH sequences encoding members of ten ALDH protein families (Jimenez-Lopez et al. 2010). Here, we modified and updated the number of ZmALDH genes in maize. ALDH proteins play essential roles in metabolic pathways that are critical for development and response to environmental changes (Kotchoni et al. 2006). The maize ALDH gene superfamily is the largest family of plant ALDHs ever characterized. A partial explanation for the presence of so many maize ALDH genes could be the need to provide ALDH activity in various subcellular compartments. The phylogenetic analysis shows that maize ALDHs split up into ten protein families (Fig. 2), which is the same as in rice (Gao and Han 2009), and confirms that these two plant species are indeed monocots. ZmALDH families 2, 5, and 10 seem to cluster together, suggesting that these families probably diverged from a common ancestor. This is in accordance with the results of recently characterized ALDH genes from Arabidopsis and rice (Kirch et al. 2004). Two genes, ZmALDH19 and ZmALDH28, were grouped to family 18. In rice, OsALDH18-1 and OsALDH18-2, encoded the P5CS (1-pyrroline-5-carboxylate synthetase) defined as ALDH-like protein (Sophos and Vasiliou 2003) with both gamma-glutamyl kinase and gamma-glutamyl phosphate reductase activities. P5CS proteins that are traditionally grouped into a distinct ALDH family (ALDH18) have great sequence divergence with proteins in other ALDH families; the most striking difference being that they do not contain the conserved ALDH active sites (Gao and Han 2009; Kotchoni et al. 2012).
Environmental stresses including drought, acid stress, and high salinity are deleterious factors for crops survival and yield. These stressors can induce the rapid and excessive accumulation of reactive oxygen species (ROS) in plant cells (Sunkar et al. 2003). The ALDH activity increase is considered as an efficient defense strategy to eliminate the toxic aldehydes caused by ROS (Rodrigues et al. 2006). Osmotic stress caused by drought, salinity, or low temperatures is a major limitation of agricultural production. Some plants synthesize and accumulate glycine betaine (GB), the most efficient osmoprotector known, when subjected to osmotic stress. In higher plants, GB is synthesized by a two-step oxidation of choline via an intermediate form of betaine aldehyde, and catalyzed by choline monooxygenase and betaine aldehyde dehydrogenase (Niu et al. 2007). Among the stress-associated ALDHs, the members of the ALDH7 family, also designated antiquitin, have not been related to any biochemical pathway. It was described as turgor responsive because the level of expression increased when the plant was dehydrated. In addition to garden pea ALDH7B1, induction of gene expression could also be observed in rapeseed ALDH7B3 under similar conditions (Stroeher et al. 1995). Antiquitin was also involved in adaptive responses mediated by a physiologically relevant detoxification pathway in plants (Rodrigues et al. 2006). These results strongly suggest that osmoprotection is a major function of antiquitin. When cells are dehydrated, osmotic stress is one of the major challenges that they have to overcome for survival. It is possible that antiquitin can oxidize some aldehyde precursors to generate the carboxylate-containing osmoprotectants. In fact, plants overexpressing ALDH9 have been reported to have significant improvement in dehydration tolerance (Ishitani et al. 1995). Microarray data analysis shows that moderate and severe drought stress repress ZmALDH9 (Fig. 3a) and ZmALDH17 (Fig. 3b) gene expression. Re-watering can rescue the expression levels of these two genes. In addition, acid soil can activate the ZmALDH13 gene expression (Fig. 4). ZmALDH9 and ZmALDH13 were classified into family 2, but ZmALDH17 was classified into family 3. Rice OsALDH2-4 was upregulated by submergence and ABA in young leaf (Tsuji et al. 2003). Previous work has established that over-expression of ALDH3I1 could improve the tolerance of transgenic plant to diverse stresses (Sunkar et al. 2003). In this quantitative real-time PCR (qRT-PCR) research, ZmALDH9, ZmALDH13, and ZmALDH17 genes showed the induced expression pattern under salt treatment, especially for ZmALDH17, which strongly induced by salt stress compared with the mock (Fig. 6b). These results indicated that families 2 and 3 genes participate in the detoxification of aldehyde generated in maize osmotic and salt stresses.
Plants are exposed to many forms of stress, one of is microbial pathogen. To effectively avoid invasion by microbial pathogens, plants have evolved sophisticated mechanisms to provide several strategic layers of constitutive and induced defenses. Recognition of pathogen-derived signal molecules by plant receptors leads to biosynthesis of one or more of the major secondary signaling molecules such as jasmonates, ethylene, and salicylic acid. These hormones then activate a series of defense responses designed to prevent further pathogen spread or plant damage (Zhou et al. 2010b). The U. maydis-maize pathosystem has emerged as the current model for plant pathogenic basidiomycetes and as one of the few models for a true biotrophic interaction that persists throughout fungal development inside the host plant. Microarray data analysis showed that ZmALDH9 and ZmALDH18 were induced after U. maydis infection (Fig. 5). Both ZmALDH9 and ZmALDH18 were classified into family 2. In the present study, ZmALDH9 was repressed at 6 and 12 h under JA treatment (Fig. 6a), which shows the opposite of what we expect. It could happen that ZmALDH9 will be induced after 12 h or more for JA treatment. The latest report shows that ectopic expression of VpALDH2B4 (Vitis pseudoreticulata) in Arabidopsis can enhance resistance to mildew pathogens and salt stress (Wen et al. 2012). Family 2 maize ALDH2B2/ RF2A was the first plant ALDH ever characterized. RF2A encodes a nuclear restorer of cytoplasmic male sterility (CMS) and functions in concert with RF1 to restore CMS in maize (Ciu et al. 1996). Here, ZmALDH9, belonging to family 2, is involved in the function of drought stress and pathogens infection. ZmALDH22 (also named as ZmALDH22A1) is induced by abiotic stresses and ABA treatment in maize seedling root. Transgenic tobacco plants overexpressing ZmALDH22A1 show elevated stress tolerance (Huang et al. 2008). The stress-induced expression patterns of ALDH genes from different model plants have been studied and it has been found that over-expression of some stress-induced ALDH genes could enhance the stress tolerance of transgenic plants. Data presented here suggested potential roles of some maize ALDH genes in the maize adaptation to environmental stressors. The present study identified 28 genes and 48 transcripts of the ALDHs family in the maize genome, and characterized ZmALDH9, ZmALDH13, ZmALDH17, and ZmALDH18 genes in response to drought, acid and U. maydis infection. The work aimed to provide a foundation for further maize breeding.
References
Bouché N, Fait A, Bouchez D, Møller SG, Fromm H (2003) Mitochondrial succinic: semialdehyde dehydrogenase of the gamma-aminobutyrate shunt is required to restrict levels of reactive oxygen intermediates in plants. Proc Natl Acad Sci USA 100:6843–6849
Ciu XQ, Wise RP, Schnable PS (1996) The RF2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 272:1334–1336
Gao C, Han B (2009) Evolutionary and expression study of the aldehyde dehydrogenase (ALDH) gene superfamily in rice (Oryza sativa). Gene 431:86–94
Huang W, Ma X, Wang Q, Gao Y, Xue Y, Niu X, Yu G, Liu Y (2008) Significant improvement of stress tolerance in tobacco plants by overexpressing a stress-responsive aldehyde dehydrogenase gene from maize (Zea mays). Plant Mol Biol 68:451–463
Ishitani M, Nakamura T, Han SY, Takabe T (1995) Expression of the betaine aldehyde dehydrogenase gene in barley in response to osmotic stress and abscisic acid. Plant Mol Biol 27:307–315
Jimenez-Lopez JC, Gachomo EW, Seufferheld MJ, Kotchoni SO (2010) The maize ALDH protein superfamily: linking structural features to functional specificities. BMC Struct Biol 10:43
Kirch HH, Bartels D, Wei Y, Schnable PS, Wood AJ (2004) The ALDH gene superfamily of Arabidopsis. Trends Plant Sci 9:371–377
Kotchoni SO, Kuhns C, Kirch HH, Bartels D (2006) Overexpression of different aldehyde dehydrogenase genes in Arabidopsis thaliana confers tolerance to abiotic stress and protects plants against lipid peroxidation and oxidative stress. Plant Cell Environ 29:1033–1048
Kotchoni SO, Jimenez-Lopez JC, Kayodé APP, Gachomo EW, Baba-Moussa L (2012) The soybeanaldehydedehydrogenase (ALDH) proteinsuperfamily. Gene 495:128–133
Mattiello L, Kirst M, da Silva FR, Jorge RA, Menossi M (2010) Transcriptional profile of maize roots under acid soil growth. BMC Plant Biol 10:196
Nicholas KB, Nicholas HB Jr, Deerfield DW II (1997) GeneDoc: analysis and visualization of genetic variation. EMBNEW News 4:14
Niu X, Zheng W, Lu B, Ren G, Huang W, Wang S, Liu J, Tang Z, Luo D, Wang Y, Liu Y (2007) An unusual posttranscriptional processing in two betaine aldehyde dehydrogenase loci of cereal crops directed by short, direct repeats in response to stress conditions. Plant Physiol 143:1929–1942
Perozich J, Nicholas H, Lindahl R, Hempel J (1999) The big book of aldehyde dehydrogenase sequences. An overview of the extended family\. Adv Exp Med Biol 463:1–7
Rodrigues SM, Andrade MO, Gomes APS, DaMatta FM, Baracat-Pereira MC, Fontes EPB (2006) Arabidopsis and tobacco plants ectopically expressing the soybean antiquitin-like ALDH7 gene display enhanced tolerance to drought, salinity, and oxidative stress. J Eep Bot 57:1909–1918
Saitou N, Nei M (1987) The neighbor–joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sophos NA, Vasiliou V (2003) zAldehyde dehydrogenase gene superfamily: the 2002 update. Chem Biol Interact 143–144:5–22
Stroeher VL, Boothe JG, Good AG (1995) Molecular cloning and expression of a turgor-responsive gene in Brassica napus. Plant Mol Biol 27:541–551
Sunkar P, Bartels D, Kirch HH (2003) Overexpression of a stress-inducible aldehyde dehydrogenase gene from Arabidopsis thaliana in transgenic plants improves stress tolerance. Plant J 35:452–464
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. doi:10.1093/molbev/msr121
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tsuji H, Meguro N, Suzuki Y, Tsutsumi N, Hirai A, Nakazono M (2003) Induction of mitochondrial aldehyde dehydrogenase by submergence facilitates oxidation of acetaldehyde during re-aeration in rice. FEBS Lett 546:369–373
Vasiliou V, Bairoch A, Tipton KF, Nebert DW (1999) Eukaryotic aldehyde dehydrogenase (ALDH) genes: human polymorphisms, and recommended nomenclature based on divergent evolution and chromosomal mapping. Pharmacogenetics 9:421–434
Wang D, Pei K, Fu Y, Sun Z, Li S, Liu H, Tang K, Han B, Tao Y (2007) Genome-wide analysis of the auxin response factors (ARF) gene family in rice (Oryza sativa). Gene 394:13–24
Wen Y, Wang X, Xiao S, Wang Y (2012) Ectopic expression of VpALDH2B4, a novel aldehyde dehydrogenase gene from Chinese wild grapevine (Vitis pseudoreticulata), enhances resistance to mildew pathogens and salt stress in Arabidopsis. Planta. doi:10.1007/s00425-012-1624-z
Zheng J, Fu J, Gou M, Huai J, Liu Y, Jian M, Huang Q, Guo X, Dong Z, Wang H, Wang G (2010) Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol Biol 72:407–421
Zhou ML, Ma JT, Pang JF, Zhang ZL, Tang YX, Wu YM (2010a) Regulation of plant stress response by dehydration responsive element binding (DREB) transcription factors. Afr J Biotechnol 9:9255–9269
Zhou ML, Zhu XM, Shao JR, Wu YM, Tang YX (2010b) Transcriptional response of the terpenoid indole alkaloid pathway to the overexpression of ORCA3 along with MeJA/nitric oxide elicitation in Catharanthus roseus hairy root culture. Appl Microbiol Biotechnol 88:737–750
Zhou ML, Hou HL, Zhu XM, Shao JR, Wu YM, Tang YX (2011) GmMYBZ2 acts as a repressor of catharanthine biosynthesis pathway in Catharanthus roseus hairy root culture. Appl Microbiol Biotechnol 91:1095–1105
Zhou ML, Zhu XM, Shao JR, Wu YM, Tang YX (2012) A Protocol for Genetic Transformation of Catharanthus roseus by Agrobacterium rhizogenes A4. Appl Biochem Biotechnol 166:1674–1684
Acknowledgments
This work was supported by a grant from the National Transgenic Program (2011ZX08005-004). We are grateful to Trillian Ross (Plant Cell Physiology, Institute of Biology, Leiden University, The Netherlands) for giving some useful comments on this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Mei-Liang Zhou and Qian Zhang contributed equally to this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 232 kb)
Rights and permissions
About this article
Cite this article
Zhou, ML., Zhang, Q., Zhou, M. et al. Aldehyde dehydrogenase protein superfamily in maize. Funct Integr Genomics 12, 683–691 (2012). https://doi.org/10.1007/s10142-012-0290-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-012-0290-3