
Abstract
Non-long terminal repeat (non-LTR) retrotransposons have contributed to shaping the structure and function of genomes. Fungi have small genomes, usually with limited amounts of repetitive DNA. In silico approach has been used to survey the non-LTR elements in 57 fungal genomes. More than 100 novel non-LTR retrotransposons were found, which belonged to five diverse clades. The present survey identified two novel clades of fungal non-LTR retrotransposons. The copy number of non-LTR retroelements varied widely. Some of the studied species contained a single copy of non-LTR retrotransposon, whereas others possessed a great number of non-LTR retrotransposon copies per genome. Although evolutionary relationships of most elements are congruent with phylogeny of host species, a new case of possible horizontal transfer was found between Eurotiomycetes and Sordariomycetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Retrotransposons are found in all investigated eukaryotes. Five orders of retrotransposons are recognized: those having long terminal repeats (LTRs; LTR retrotransposons), those lacking LTRs (non-LTR retrotransposons), DIRS retrotransposons, Penelope-like retrotransposable elements, and short interspersed nuclear elements (SINEs; Finnegan 1992; Wicker et al. 2007). Several enzymatic activities can be distinguished in proteins encoded by functional non-LTR retrotransposons (Malik et al. 1999). The key component is reverse transcriptase (RT) encoded by all non-LTR retrotransposons. The second component is endonuclease, which is encoded by restriction-enzyme-like endonuclease (REL-endo) domains in some elements and by apurinic/apyrimidinic (APE) endonuclease in others. The first ORF, if present, encodes a gag-like protein with a function of nucleic acid chaperone (Martin and Bushman 2001). Finally, some elements also contain ribonuclease H (RNH) domains.
Phylogenetic analysis of non-LTR retrotransposons based on the reverse transcriptase domains allowed distinguishing 21 clades (Malik et al. 1999; Malik and Eickbush 2000; Volff et al. 2000; Lovsin et al. 2001; Arkhipova and Morrison 2001; Burke et al. 2002; Biedler and Tu 2003). Based on structural and phylogenetic features of different elements, Malik et al. (1999) developed a scenario for evolution of non-LTR retrotransposons and demonstrated that non-LTR retrotransposons are inherited mostly by vertical transmission. Only a few cases of possible horizontal transfer of non-LTR retrotransposons have been described (Župunski et al. 2001; Sánchez-Gracia et al. 2005; Novikova et al. 2007).
The most ancient clades of non-LTR retrotransposons (GENIE, CRE, R2, NeSL-1, and R4) contain only one ORF and show site-specific distribution in the genomes (Malik et al. 1999; Malik and Eickbush 2000). They have domain of restriction-enzyme-like endonuclease (REL-endo). During further evolution of mobile elements, the REL-endo domain is suggested to have been substituted with an apurinic/apyrimidinic (APE) endonuclease acquired from the host cells. All younger clades (L1, RTE, Tad, R1, LOA, I, Jockey, CR1, Rex1, and L2) possess the APE endonuclease domain and are called APE retrotransposons (Zingler et al. 2005). The acquisition of the APE endonuclease resulted in a loss of target site specificity for all the elements (except R1 clade and some elements from L1 clade) and coincided with the origin of a second ORF in front of the RT-encoding ORF. Finally, elements of some clades obtained one more enzymatic domain in the second ORF, the RNH domain.
RNH domain appears to be a much more ancient acquisition of non-LTR retrotransposons than proposed earlier, subsequently lost by the majority of younger non-LTR retrotransposons (Malik et al. 1999; Kojima and Fujiwara 2005). For a long time, RNH domain was detected only in younger clades of non-LTR retroelements such as Tad, R1, LOA, and I (Malik et al. 1999; Malik and Eickbush 2001; Malik 2005). It was therefore believed that RNH domain was acquired later than APE endonuclease domain. However, a recent description of Dualen elements from Chlamydomonas reinhardtii suggests that RNH domain appeared much earlier in the evolution of non-LTR retrotransposons (Kojima and Fujiwara 2005). Dualen elements are believed to have appeared before L1 clade and have single ORF which encodes a polyprotein that has REL-endo, RT, APE, and RNH domains.
Fungi have small genomes, usually with limited amounts of repetitive DNA. Among the Eumycota, the younger evolutionary divisions, Ascomycota and Basidiomycota, have a strong tendency towards streamlined genomes. Representatives of Eumycota contain not more than 10–15% of repetitive DNA, including retrotransposons (Kempken and Kück 1998; Wöstemeyer and Kreibich 2002). Only three clades of non-LTR retrotransposons are known in fungi: Tad, L1, and CRE. Tad clade is a completely fungal group, which for a long time was believed to be the sole group of non-LTR retrotransposons represented in fungi (Malik et al. 1999). Several Tad clade elements were described from the model objects Neurospora crassa and Magnaporthe grisea (Kinsey and Helber 1989; Hamer et al. 1989); CgT1 was described from Colletotrichum gloeosporioides, and Mars1, from Ascobolus immersus (He et al. 1996; Goyon et al. 1996). Whole genomic sequences analysis of dimorphic yeasts Yarrowia lipolytica (Ylli element) and Candida albicans (Zorro element) showed that fungal non-LTR retrotransposons are not limited to Tad clade. L1 clade elements were described from the genomes of these yeasts (Goodwin et al. 2001; Casaregola et al. 2002). L1-like element was also described from a basidiomycete Microbotryum violaceum and a glomeromycete Gigaspora (Hood 2005; Gollotte et al. 2006). Finally, element Cnl from Cryptococcus neoformans was found to belong to the ancient CRE clade (Goodwin and Poulter 2001).
Non-LTR retrotransposons survey from genomic sequences
Fungal genomic sequences are available at: Fungal Genome Initiative (Broad Institute: http://www.broad.mit.edu/annotation/fgi/), The DOE Joint Genome Institute (JGI: http://www.jgi.doe.gov/), Génolevures (http://cbi.labri.fr/Genolevures/), The Sanger Institute (http://www.sanger.ac.uk/). The source of individual genomes could be found in Table S1 of Supplementary materials. All downloads were performed before 1 May 2008.
We used UniPro GenomeBrowser software (http://genome.unipro.ru/) for non-LTR retroelements identification. The investigated genomes were translated over six possible reading frames in protein form, on which search of homologous regions was performed using “HMMER search” options of UniPro GenomeBrowser. The algorithm of HMMER search is based on profile hidden Markov models, which can perform amino acid sequence searches by use of the appropriate profile (McClure et al. 1996; Eddy 1998).
For the analysis, we used a multiple alignment consensus sequence, which contains information about RT domain. The profile HMM, based on this consensus sequence, was also built using UniPro GenomeBrowser software. Such models are constructed with position-specific scores for amino acids and position-specific penalties for opening and extending an insertion or deletion, and represent a statistical description of a certain multiple alignment. Profile HMMs can be used for searching for additional remote homologues of the sequence family. An additional check for the presence of RT domain was performed using BLAST analysis. BLAST was essentially performed using sequence databases accessible from the National Center for Biotechnology Information (NCBI) server (www.ncbi.nlm.nih.gov/BLAST/). The classification of the newly identified elements was performed by comparative analysis of their sequences. Based on the observed sequence divergence distribution, all sequences obtained from the same fungal genome were referred to as copies of one element if they shared similarity more than 90%. Newly identified elements and their accession numbers in public databases are listed in Supplementary material Table S2.
The nucleotide sequences of the elements were also extracted with the assistance of UniPro GenomeBrowser software. After localization of amino acid sequences obtained during HMMER search in the initial genomes in its nucleotide representation, the sequences were expanded up to the 10 kb and used for multiple alignments with other copies of the same element. The visualization feature and “ORF Find” option of UniPro GenomeBrowser were used to identify the putatively intact copies of non-LTR retrotransposons.
Multiple DNA alignments were performed by ClustalW (Thompson et al. 1994) and edited manually. Phylogenetic analyses were performed using the neighbor-joining (NJ) method in MEGA 4.0 program (Tamura et al. 2007). Statistical support for the NJ tree was evaluated by bootstrapping (number of replications, 1,000; Felsenstein 1985).
Evolutionary rates were estimated by standard methods (Nei and Kumar 2000). Poisson correction distances (d) were estimated from the equation d = −ln(1 − p), where p represents the proportion of different amino acids. The rate of amino acid substitution (r) was estimated by the standard equation r = d/2T, where T is the divergence time of the last common ancestor (LCA) of the compared species. Amino acid distances used in divergence-versus-age analysis were calculated from sequences of the RT domain using MEGA 4.0 (Tamura et al. 2007).
Identification and classification of non-LTR retrotransposons in Fungi
A total of 57 species were included in our analysis. The list of analyzed fungal species, their taxonomy, genomic size, and results of in silico search are presented in Table 1. The majority of investigated fungi gave positive results during in silico search of non-LTR retrotransposons. However, some species have no non-LTR retrotransposons. Investigated representatives of the phylum Microsporidia (Antonospora locustae and Encephalitozoon cuniculi) did not possess detectable non-LTR retrotransposons (Table 1). Microsporidian fungi are obligate intracellular eukaryotic parasites, which lack typical eukaryotic organelles, have small ribosomes (Cavalier-Smith 1991), and extremely small genomes, only 2.5–3 Mb (Peyretaillade et al. 1998; Vivarès and Méténier 2000). It is not surprising that studied microsporidians lack repeated sequences such as non-LTR retrotransposons.
Majority of the investigated saccharomycetes did not have non-LTR retrotransposons. Absence of non-LTR retrotransposons was reported previously for yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe (Jordan and McDonald 1999; Wood et al. 2002). Only several Saccharomycotina species showed the presence of non-LTR retrotransposons in their genomes (Pichia stipitis CBS 6054, present study; C. albicans, Goodwin et al. 2001; and Y. lipolytica, Casaregola et al. 2002). Non-LTR retrotransposons also were not found in a few sordariomycetes (Fusarium graminearum PH-1 and Trichoderma reesei QM6a), heterobasidiomycetes (Malassezia globosa CBS 8777), and chytridiomycetes (Batrachochytrium dendrobatidis JEL423) (Table 1). This could be due either to current status of genome sequencing or to distribution of non-LTR retrotransposons in chromosomes. The pericentromeric and subtelomeric regions, which are enriched by repeated elements, remain unfinished during sequencing processes and genome assembly (Eichler et al. 2004; Galagan et al. 2005). It is also highly possible that these species lack non-LTR retrotransposons.
In total, 32 fungal genomes gave positive signals in HMM search of non-LTR retrotransposons. The number of putative non-LTR retrotransposons varies considerably from species to species (Table 1). Identified sequences of non-LTR retrotransposons were classified based on their intraspecific similarity for each species and were either highly similar or very different. All amino acid sequences with similarity more than 90% were considered to be products of the copies of the same element. Totally, 130 novel non-LTR retrotransposons were found (see Supplementary material Table S2 and Table S3). The non-LTR retrotransposon sequences do not exceed 2% in investigated fungal genomes. In fact, in the majority of investigated species, non-LTR retrotransposons comprise not more than 0.5% of the entire genome (Supplementary material Fig. S1).
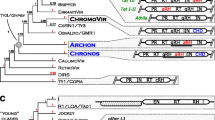
Preliminary phylogenetic analysis showed that all identified non-LTR retrotransposons fall into five distinct lineages. The overwhelming majority of the elements belong to Tad clade; the rest could be referred to the L1 and CRE as well as to previously unknown clades (Fig. 1).

Neighbor-joining (NJ) phylogenetic trees based on RT amino acid sequences of non-LTR retrotransposons including newly described Tad-like elements (a), and elements belonged to the L1 and RTE clades as well as newly identified clades Inkcap and Deceiver from fungi (b). Statistical support was evaluated by bootstrapping (1,000 replications); nodes with bootstrap values over 50% are indicated. The name of the host species and accession number are indicated for the elements taken from GenBank. The clades (bold) and families (italics) are shown on the right. Elements AniNLR2 and AorNLR2, which possibly was horizontally transmitted, are italicized and underlined. Novel clades, Deceiver and Inkcap, are marked
Sequence diversity and structure of Tad-like elements
Elements from Tad clade were found in all studied species which showed the presence of non-LTR retrotransposons, except yeasts, C. neoformans (Heterobasidiomycota), and Sporobolomyces roseus (Urediniomycota). Nine distinct families could be recognized inside Tad clade on the phylogenetic tree based on RT domain (Fig. 1a).
Families of Tad-like non-LTR retrotransposons appeared to be specific for either Ascomycetes or Basidiomycetes fungi. Ascomycetes were represented by six families (Mars1, CgT, Tad1, Ask1, Ask2, and Ask3), whereas the Basidiomycetes, only by three (But1, But2, and But3; Fig. 1a).
Our analysis showed that each investigated fungal species has a unique number of non-LTR retrotransposon families from Tad clade. For example, five diverse families are represented in the genome of Histoplasma capsulatum NAm1, whereas a single family was found in both Aspergillus niger ATCC1015 (Ascomycota) and Coprinus cinereus Okayama7#130 (Basidiomycota). Six different non-LTR retrotransposons were detected in the genome of Chaetomium globosum CBS 148.51, which belonged to two families, Ask1 and Tad1.
For each distinct Tad-like non-LTR retrotransposon, we attempted to isolate a representative full-length sequence. For the purpose of our analysis, a “full-length element” is defined as one that has two recognizable open reading frames (ORF). The majority of elements appeared to be degenerate in investigated fungal genomes (see Supplementary material Table S3). Nevertheless, we have significantly increased the number of novel full-length non-LTR retrotransposons known in Ascomycetes and, especially, in the Basidiomycetes.
Newly detected full-length and intact fungal retrotransposons from Tad clade are approximately 6,000 bp in length (Fig. 2). First, ORF as a rule encodes a nucleic acid-binding protein with cysteine motif of the CX2CX4HX4C type (CCHC type). Second ORF encodes a polyprotein (ORF2p) with AP endonuclease and RT domains. ORF2p from majority of detected elements have an additional ribonuclease H domain (RNH) downstream of the RT domain and a cysteine motif of CCHC type.

Structural organization of the fungal full-length elements in Tad, L1, Deceiver, Inkcap, and CRE clades. ORF1 First open-reading frame, ORF2 second open reading frame, RT reverse transcriptase, RH ribonuclease H, AP apurinic/apyrimidinic endonuclease, REL-endo restriction-enzyme-like endonuclease, CCHC and CCHH Zn-finger motifs, ND not determined, asterisk stop codon
The ribonuclease H domain in Tad clade
Not all newly described fungal retroelements showed the presence of RNH enzymatic domain. Representatives of two families (Tad1 and Ask3) and some of the elements from family Ask1 have no detectible RNH (Fig. 2; Supplementary material Table S3). Elements from Tad1 and Ask3 families seem to have been evolved from a common ancestor, which lacked RNH. No traces of this domain were found during our analysis. The Ask1 family is unique among fungal families from Tad clade. Some of the Ask1 elements lack RNH (AorNLR2, AniNLR2, FoNLR5-FoNLR8, CgNLR2-CgNLR4), whereas others have it (HcNLR5, AorNLR3, AorNLR4, AniNLR3 and AniNLR4).
Elements from C. globosum and Fusarium oxysporum, which belong to Ask1, were analyzed for the traces of RNH in their sequences. We found the evidence that RNH domain was lost from FoNLR5, FoNLR6, FoNLR8, CgNLR2, CgNLR3, and CgNLR4 elements (Fig. 3). The traces of RNH domain were determined for the ORF2p C-terminal sequence in FoNLR5, FoNLR6, FoNLR8, CgNLR2, and CgNLR4. One of the RNH catalytic motifs (key residue is E48) could be clearly recognized on the multiple alignment presented in Fig. 3. However, residues D10, D70, H124, and D134, which are believed to be essential for the enzymatic activity of the RNH, were absent (Kanaya et al. 1996; Malik and Eickbush 2001).

Alignment of the ribonuclease H (RNH) domains. Representative RNH domains from Eukarya (including Fungi and Metazoa), non-LTR retrotransposons Lian from Aedes aegypti (GenBank acc. No. U87543), Bilbo from Drosophila subobscura (GenBank acc. No. U73800), CgT1–3 from Glomerella cingulata (GenBank acc. No. L76169), and Tad-like non-LTR retrotransposons from diverse families were aligned using ClustalW. Conserved residues are highlighted in bold; those which believed to be important for the catalytic mechanism of RNH are indicated by arrows, including the four carboxylate (dark arrows) and the single histidine residue (white arrow) that are numbered according to their position in the Escherichia coli RNH domain
CgNLR3 elements have a shorter ORF2 in comparison with CgNLR2 and CgNLR4 and other full-length intact elements from Ask1 family. It seems that RNH domain was lost in CgNLR3 element as a result of stop codon appearance which cut off the enzymatic domain. We analyzed nucleotide sequences of all four CgNLR3 copies. All of them had this stop codon at the same position. Thus, existence and activity of self-coding RNH domain appear not to have been crucial for recent retrotransposition of CgNLR3. It remains unclear why elements from C. globosum and F. oxysporum lack RNH.
The copies of CgNLRs (CgNLR2-CgNLR4) and FoNLR5 retrotransposons shared a very high similarity at the DNA level. The majority have intact ORFs with flanking target site duplications. It seems that CgNLRs and FoNLR5 were recently active, and the absence of RNH did not affect their transposition. Since the TPRT mode of transposition involves reverse transcription at the future insertion site in genomic DNA, the non-LTR retrotransposons have access to the host-encoded RNH activity (Eickbush and Malik 2002; Malik 2005). We could suggest that activity of cellular RNH is sufficient for successful retrotransposition of non-LTR retrotransposons in C. globosum and F. oxysporum.
AniNLR2 from A. niger and AorNLR2 from Aspergillus oryzae also had only a degenerate RNH domain, whereas AniNLR4, AorNLR3, and AorNLR4 elements from the same family possessed RNH (Fig. 3). Moreover, AniNLR2 and AorNLR2 appeared to be more closely related to the non-LTR retrotransposons from C. globosum and F. oxysporum than to those from Aspergillus fumigatus (AfNLR2) and other elements from A. oryzae and A. niger. AniNLR2 and AorNLR2 non-LTR retrotransposons exhibit an unexpectedly high similarity with elements FoNLRs and CgNLRs. For example, RT regions of AorNLR2 and CgNLR3 were more than 88% similar in their amino acid sequences and 71.3% similar at the DNA level. At the same time, RT domain of AorNLR2 showed the average similarity of only 59% with AorNLR3 and AorNLR4. This could be explained either by strong selective constraints in RT sequence coupled with a strict vertical transmission or by horizontal transmission.
Possible horizontal transmissions of Tad-like retroelements in fungi
Most examples of horizontal transfer (HT) of eukaryotic genes involve transposable elements (Kidwell 1992; Hartl et al. 1997; Jordan et al. 1999). Such transfers are usually recognized by the presence of very closely related elements in distant host taxa. Although HT is well known for LTR retrotransposons and especially for DNA transposons (Robertson 1993; Silva and Kidwell 2004), non-LTR retrotransposons rarely undergo HT, and their phylogenies are largely congruent to those of their hosts (Malik et al. 1999). There are a few cases of well-documented HT for non-LTR retrotransposons: (a) HT of Bov-B elements (RTE clade) from the ancestral snake lineage (Boidae) to the ancestor of ruminant mammals, dated 40–50 Mya (Kordiš and Gubenšek 1998), (b) HT of CR1 elements between Maculinea butterflies and Bombyx moths, dated 10–20 Mya (Novikova et al. 2007), and (c) HT of non-LTR retrotransposons in Drosophila melanogaster that probably occurred ~5–12 million years ago (Sánchez-Gracia et al. 2005). Other examples of HT were demonstrated for Jockey elements (Mizrokhi and Mazo 1990) and CR1 elements (Drew and Brindley 1997).
Using the criterion of divergence of functional proteins and proteins encoded by transposable elements, evidence for HT was obtained. In the case of lower divergence among mobile elements than that observed for the proteins encoded by host genes, very strict evolutionary constrain or HT could be proposed as explanation (Sánchez-Gracia et al. 2005; Novikova et al. 2007). The analysis of amino acid sequence divergences was performed for non-LTR retrotransposons belonging to the Ask1 family and the cellular proteins from A. fumigatus, A. niger, A. oryzae, F. oxysporum, and C. globosum. The RT domain of non-LTR retrotransposons appeared to be more diverse than the compared cellular proteins with the exception of AorNLR2/CgNLR3, AorNLR2/FoNLR5, AniNLR2/CgNLR3, and AniNLR2/FoNLR5 pairs (Table 2). On the one hand, RT domains of AniNLR2, AorNLR2, CgNLRs, and FoNLRs are more divergent than the most conservative proteins (e.g., elongation factor, EF-1; Table 2). On the other hand, they showed higher similarity than the majority of the other investigated proteins (e.g., adenylate kinase Adk; Table 2; see Supplementary materials Table S4).
The slowdown effect on evolutionary rates accompanied the previously described possible HT cases (Župunski et al. 2001; Sanchez-Gracia et al. 2005; Novikova et al. 2007; Roulin et al. 2008). We estimated the evolutionary rates for fungal non-LTR retrotransposons from Tad clade and compared them with early calculated evolutionary rates for invertebrate CR1- and Jockey-like non-LTR retrotransposon and vertebrate RTE-like non-LTR retrotransposons (Table 3; Župunski et al. 2001; Novikova et al. 2007). In general, the evolutionary rates in fungal retrotransposons are higher than those observed in metazoan non-LTR retrotransposons. At the same time, comparisons of AniNLR2, AorNLR2, CgNLR3, and FoNLR5 retrotransposons demonstrated significantly lower evolutionary rates than Tad non-LTR retrotransposons in all other comparisons (Table 3).
The HT nature of the mobile elements was further tested by the divergence-versus-age analysis (Malik et al. 1999; Kordiš and Gubenšek 1998). It includes the comparison of divergence rates between the RT domains of the non-LTR retrotransposons with the host divergence time estimates. Amino acid sequence distances between the RT domains of the Tad clade representatives along with other elements (from R1, R2, Jockey, I, CR1, RTE, and L1 clades) were plotted against estimates of host divergence time (Fig. 4). The time since divergence of Eurotiomycetes and Sordariomycetes is estimated between 310 and 670 Myr (Berbee and Taylor 1993; Heckman et al. 2001). The oldest well-documented ascomycete fossils are found in the 400-Myr-old Rhynie chert (Taylor et al. 1999). Based on this finding, it was proposed that 400 Myr for the Eurotiomycetes and Sordariomycetes divergence would seem to provide a conservative date estimate; however, earlier dates could be expected (Kasuga et al. 2002). We used the date 400 Myr for the last common ancestor of Eurotiomycetes and Sordariomycetes for further analysis.

Divergence-versus-age analysis of the non-LTR retrotransposons including Tad clade. Amino acid sequence distances were calculated from the sequences of the complete reverse transcriptase (RT) domain. The curves for arthropods and vertebrates are reproduced from Malik et al. (1999). For each host divergence time estimate, the fungal elements used are as follows: CiNLR1 versus UrNLR1 (1) compared at 25 Myr; AniNLR1 versus AfNLR2 (2), at 44 Myr; AniNLR3 versus AfNLR2 (3), at 44 Myr; Tad versus CgNLR1 (4), at 70 Myr; CcNLR1 versus LbNLR6 (5), at 90 Myr; Tad versus HcNLR6 (6), at 400 Myr; CcNLR1 versus PcNLR1 (7), at 180 Myr; CgNLR3 versus AniNLR3 (8), at 400 Myr; LbNLR1 versus Mars (9), at 1,200 Myr; AniNLR2 versus CgNLR3 (10), at 400 Myr; AniNLR2 versus FoNLR5 (11), at 400 Myr; AorNLR2 versus CgNLR3 (12) at 400 Myr; AorNLR2 versus FoNLR5 (13), at 400 Myr. Species divergence times are based on estimates by Bowman et al. (1996) for Coccidioides and Uncinocarpus comparison, by Kasuga et al. (2002) for comparisons between Eurotiomycetes and Sordariomycetes as well as between A. niger and A. fumigatus, Hibbett et al. (1997) for Homobasidiomycetes, and by Heckman et al. (2001) for comparisons between Ascomycetes and Basidiomycetes. HT events described earlier are also indicated: Bov-B elements Bos taurus (Bta) and Vipera ammodytes (Vam); CR1B elements from Bombyx mori (BmCR1B) and Maculinea teleius (MteCR1B)
Amino acid sequence distances versus host divergence time were compared within and between basidiomycete and ascomycete Tad lineages (Fig. 4). The estimated time divergence between these two groups of fungi was 1,210 Myr (Hedges 2002). Almost all ascomycete comparisons fall above the arthropod and vertebrate curves, suggesting that non-LTR sequence evolution in ascomycete fungi is faster than that in arthropods and vertebrates. The intergroup Tad comparisons, Laccaria versus Ascobolus and Coprinus versus Ascobolus, fall near the arthropod curve. However, comparisons of taxa separated by more than 600 Myr have low resolution (Malik et al. 1999; Župunski et al. 2001). All basidiomycete comparisons also fall near the arthropod curve that indicates similar rates of transposable element evolution in basidiomycetes and arthropods. Several examples, in which the points fell markedly below all curves, were AniNLR2 versus CgNLR3, AorNLR2 versus CgNLR3, AniNLR2 versus FoNLR5, and AorNLR2 versus FoNLR5 (points 10–13, at 400 Myr). It could be explained by a HT event or very strict evolutionary constraints. A HT event is more likely, since the selective pressure could be implemented only in the case of functional importance of the AorNLR2 and AniNLR2 insertions. It is known that the insertion of a transposable element can alter gene expression and be selectively advantageous. However, only part of such transposable elements evolves under selective pressure (Medstrand et al. 2001; Ono et al. 2001). We did not find any evidence that AorNLR2 and AniNLR2 elements or their parts are involved in the domestication processes. Additionally, horizontal transfer seems to be very important part of the fungal evolutionary history. HT is considered to be a key event in the evolution of several fungal genes (Wenzl et al. 2005; Slot and Hibbett 2007; Khaldi et al. 2008).
L1 and CRE clades in fungal genomes
New representatives of L1 clade were found in the yeast P. stipitis CBS 6054 and in five investigated Basidiomycetes: C. cinereus Okayama7#130, Laccaria bicolor S238N, Ustilago maydis 521, Postia placenta MAD-698, and Phanerochaete chrysosporium RP-70 (Fig. 1b).
Elements from P. stipitis CBS 6054 clustered together with Zorro3 element from the yeast C. albicans and Ylli element from Y. lipolytica (Goodwin et al. 2001; Casaregola et al. 2002). All PsNLRs are intact non-LTR retrotransposons, which have two ORFs and a polyA tail typical for L1 clade elements (Fig. 2; Supplementary material Table S3). The ORF2p of PsNLRs showed presence of both AP and RT domains. We compared ORF2p from newly isolated PsNLRs to the known elements from other yeasts (Zorro and Ylli). PsNLR1 and Zorro3 elements demonstrated 37.8% amino acid sequence similarity, whereas PsNLR1 and other PsNLRs showed only 25.5% similarity. PsNLR2, PsNLR3, and PsNLR4 are closely related and have more than 60% identical amino acid residues. It is interesting that the majority of PsNLRs copies in P. stipitis genome were intact, full-length elements, whereas other studied fungi contained predominantly degenerate copies of non-LTR retrotransposons (Supplementary material Table S3).
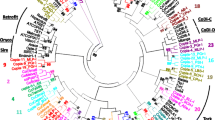
L1-like retroelements from Basidiomycetes formed a separate cluster (Fig. 5). Both phylogenies (Bayesian inference and neighbor-joining) did not resolve the position of UmNLR1. UmNLR1 is a single, full-length, intact non-LTR retrotransposon detected in genome of U. maydis 521 (Fig. 2). It has an ORF1, typical for L1 clade. The protein product of UmNLR1 ORF1 carries two cysteine motifs, which have CCHC composition. ORF2p has AP and RT domains as well as CCHC motif at its C terminus.

CcNLR6 from C. cinereus Okayama7#130, LbNLR9 from L. bicolor S238N, PcNLR7 from P. chrysosporium RP-70, and three elements from P. placenta MAD-698 (PpNLR4, PpNLR5 and PpNLR6) all seem to be closely related non-LTR retrotransposons. They share more than 66% of similarity of the RT domain amino acid sequences. None of these elements was a full-length intact non-LTR retrotransposon. CcNLR6 has a putatively intact ORF2 which encodes a protein with AP and RT enzymatic domains. An additional short ORF also was found upstream, but its protein product did not show any features of a retrotransposable ORF1p. CcNLR6 was located at the 3′ end of the supercontig 1.32 (GenBank Acc. No AACS01000351) in a reverse orientation. It could be possible that after final release of complete C. cinereus Okayama7#130 genome, CcNLR6 will be completely reconstructed. Pseudo-ORF2 could be found in PpNLR4 non-LTR retrotransposon from P. placenta. This retrotransposon like CcNLR6 possesses additional short putative ORF1 upstream (Fig. 2). However, the protein product of this additional ORF shares a very low similarity with ORF1p from CcNLR6 and did not show any presence of functional protein domains. Origin and function of ORF1s from CcNLR6 and PpNLR4 remain unclear.
Two non-LTR retrotransposons, FvNLR4 from Fusarium verticillioides 7600 and FoNLR9 from F. oxysporum 4286 FGSC, appeared to be closely related to the Cnl element from C. neoformans, which belonged to the CRE clade (Goodwin and Poulter 2001; Fig. 1b). The newly detected FvNLR4 and FoNLR9 are the first CRE-like non-LTR retrotransposons from Ascomycetes. FvNLR4 is a highly degenerate retrotransposable element. Nevertheless, RT domains of Cnl and FvNLR4 elements showed 36.8% similarity. At the same time, FoNLR9 retrotransposon is presented by two putatively intact copies per genome and possesses single ORF (Fig. 2). The FoNLR9s ORF is 3,435 bp in length and encodes a protein with RT domain and restriction-enzyme-like endonuclease (REL-endo) domain containing the CCHC motif and located downstream from RT. Two additional CCHH cysteine motifs were found at the N-terminal end of protein (Fig. 2).
Novel clades of fungal non-LTR retrotransposons
The reconstructed phylogenetic trees revealed the presence of two additional groups of elements. Malik et al. (1999) proposed to use the term “clade” to represent non-LTR retrotransposons that are grouped together with high phylogenetic support, share the same structural features, and date back to the Precambrian era (older than ~570 Myr). The newly identified groups satisfy these terms. They have strong phylogenetic support in both neighbor-joining and Bayesian inference trees and cannot be referred to the known clades (Fig. 1b and Supplementary material Fig. S2). Both groups appeared more than 900 Mya before the divergence of Uredinomycetes and Hymenomycetes (Hedges 2002).
Three newly described retrotransposons CcNLR7, LbNLR8, and PgtNLR7 (from Puccinia graminis f. sp. tritici) formed a clade which was named Deceiver (Fig. 1b). They showed 60% average similarity of the RT domains at the amino acid level. CcNLR7 is represented by two copies per genome of C. cinereus Okayama7#130. They were located in the supercontig 1.55 (GenBank Acc. No AACS01000377) and represented a nested insertion of one copy into another. One of the ORFs was reconstructed based on these two copies. It carried typical AP and RT domains. LbNLR8 from L. bicolor S238N is a full-length, putatively intact non-LTR retrotransposon represented by a single copy per genome (Fig. 2). LbNLR8 has two open reading frames: ORF1 encodes protein with a cysteine motif of CCHC type; product of ORF2 showed the presence of AP and RT domains. PgtNLR7 non-LTR retrotransposon is also represented by single copy. Only pseudo-ORF could be reconstructed in internal part of PgtNLR7 (Supplementary material Table S3). The clade branching just before Deceiver is the L1 clade, and the clade branching just after it is the RTE clade. The Bayesian inference did not resolve relationships between Deceiver and RTE clades (Supplementary material Fig. S2).
Novel clade named Inkcap was found in the genomes of C. cinereus Okayama7#130 and Sporobomyces reseus. Three non-LTR retrotransposons belonged to this clade, CcNLR4, CcNLR5, and SrNLR1. All of them were degenerate non-LTR retrotransposons. Nevertheless, a relatively long ORF could be reconstructed for SrNLR1 (Fig. 2). CcNLR4, CcNLR5, and SrNLR1 shared 60% average similarity at the amino acid level. It seems to be that Inkcap appeared just after RTE and Deceiver clades (Fig. 1b).
Distribution of diverse clades in Fungi
Five distinct clades were found in fungal genomes. Non-LTR retrotransposons from Tad clade were identified in almost all investigated species. It is widely distributed and appeared at least before Basidiomycota and Ascomycota divergence. Later on, Tad-like elements were lost by a common ancestor of Saccharomycetes. None of the 17 investigated species from Saccharomycotina lineage have non-LTR retrotransposons that belonged to the Tad clade (Fig. 5).
The novel elements from L1 clade were described from genomes of Basidiomycota and yeast P. stipitis. It is interesting that L1 clade was not found in the non-yeast Ascomycetes. L1 clade is one of the most widely distributed clades. L1-like non-LTR retrotransposons was described for all eukaryotic groups: Protista, Plantae, Fungi, and Metazoa (Malik et al. 1999; Goodwin et al. 2001; Casaregola et al. 2002; Zingler et al. 2005). Nevertheless, it seems that L1 clade is represented only in a few fungal groups such as Glomeromycetes (Gigaspora, Gollotte et al. 2006), Homobasidiomycetes, and Ustilaginomycetes from Basidiomycota and a number of Saccharomycotina species from Ascomycota. L1 clade was completely lost by Eumycota fungi (Fig. 5).
The CRE clade elements were found in two Fusarium species. CRE clade is one of the oldest clades of non-LTR retrotransposons (Malik et al. 1999). Initially, representatives of the CRE clade were found in the genomes of Trypanosomatidae (Protista: Kinetoplastida; Teng et al. 1995; Aksoy et al. 1990). Recently, CRE-like non-LTR retroelement was described from the genome of an encapsulated yeast C. neoformans (Goodwin and Poulter 2001). A sporadic distribution of this clade indicates that some fungi retained ancient non-LTR retrotransposons, obtained from their last common ancestor with protists, but majority of investigated species have lost CRE-like elements. A comprehensive survey of repeated elements from diverse fungal species could further increase the number of representatives of CRE clade.
Finally, two previously unknown clades were described in Basidiomycota, Deceiver (from three species), and Inkcap (from two species). It is highly possible that Deceiver and Inkcap have a limited distribution among fungi; their phylogenetic status and distribution require further examination (Fig. 5).
Evolutionary dynamics of non-LTR retrotransposons in fungi
The results of our survey of non-LTR retrotransposons from 57 fungal genomes showed that the copy number and percentage of non-LTR retroelements per genome varied widely (Table 1; see Supplementary material Table S3 and Fig. S1). Some of the investigated species contained single copy (e.g., Botrytis cinerea B05.10), whereas others possessed a great number of non-LTR retrotransposon copies per genomes (e.g. C. globosum CBS 148.51). It is clear that diversity of non-LTR retrotransposons and their copy number depends on the evolutionary history of a particular species or a cluster of closely related species, their population structure as well as ecological aspects.
There are several main processes which could affect the copy number and diversity of non-LTR retrotransposons in fungal genomes: stochastic loss of non-LTR retrotransposons, burst of retrotransposition, the limitation of copy number increase by natural selection which removes deleterious insertions, horizontal transfer, passive and active inactivation of repetitive sequences, self-regulation of transposition (decrease of the transposition rate when the copy number increases; e.g. Hua-Van et al. 2005; Le Rouzic and Capy 2005; Johnson 2007). The population structure and dynamics as well as mating mode also play an important role in the transposable elements evolution (Arkhipova 2005; Johnson 2007).
Those species, which have only several copies of non-LTR retrotransposons per genome, could lose these elements as a result of genetic drift, especially if the population is small and non-LTR retrotransposons are represented only by degenerate copies (Brookfield and Badge 1997). On the other hand, if the non-LTR retrotransposon is presented by at least one intact copy capable for retrotransposition, it could invade a population assuming that its transposition activity counterbalances its loss due to natural selection (Hickey 1982; Le Rouzic and Capy 2005). The inactivation of repeated sequences is also a very important factor, which leads to the shifts in diversity and copy number of non-LTR retrotransposons, especially in fungi. Existence of diverse strategies countering the short-term spreading of repetitive elements is known for fungi: they include methylation induced premeiotically (MIP), repeat-induced point mutation (RIP), and quelling (Faugeron 2000; Cogoni and Macino 1999; Galagan and Selker 2004). The complex interactions between various forces lead to the formation of unique repertoire of non-LTR retrotransposons in each fungal species.
References
Aksoy S, Williams S, Chang S, Richards FF (1990) SLACS retrotransposon from Trypanosoma brucei gambiense is similar to mammalian LINEs. Nucleic Acids Res 18:785–792
Arkhipova IR (2005) Mobile genetic elements and sexual reproduction. Cytogenet Genome Res 110:372–382
Arkhipova IR, Morrison HG (2001) Three retrotransposon families in the genome of Giardia lamblia: two telomeric, one dead. Proc Natl Acad Sci USA 98:14497–14502
Berbee ML, Taylor JW (1993) Dating the evolutionary radiations of the true fungi. Can J Bot 71:1114–1127
Berbee ML, Taylor JW (2001) Fungal molecular evolution: Gene trees and geologic time. In: McLaughlin DJ, McLaughlin EG, Lemke PA (eds) The mycota: a comprehensive treatise on fungi as experimental systems for basic and applied research. Volume VII: Systematics and Evolution, Part B, (2001). Springer-Verlag, New York
Biedler J, Tu Z (2003) Non-LTR retrotransposons in the African malaria mosquito, Anopheles gambiae: unprecedented diversity and evidence of recent activity. Mol Biol Evol 20:1811–1825
Bowman BH, White TJ, Taylor JW (1996) Human pathogeneic fungi and their close nonpathogenic relatives. Mol Phylogenet Evol 6:89–96
Brookfield JF, Badge RM (1997) Population genetics models of transposable elements. Genetica 100:281–294
Burke WD, Malik HS, Rich SM, Eickbush TH (2002) Ancient lineages of non-LTR retrotransposons in the primitive eukaryote, Giardia lamblia. Mol Biol Evol 19:619–630
Casaregola S, Neuveglise C, Bon E, Gaillardin C (2002) Ylli, a non-LTR retrotransposon L1 family in the dimorphic yeast Yarrowia lipolytica. Mol Biol Evol 19:664–677
Cavalier-Smith T (1991) Archamoebae: the ancestral eukaryotes? Biosystems 25:25–38
Cogoni C, Macino G (1999) Homology-dependent gene silencing in plants and fungi: a number of variations on the same theme. Curr Opin Microbiol 2:657–662
Drew AC, Brindley PJ (1997) A retrotransposon of the non-long terminal repeat class from the human blood fluke Schistosoma mansoni. Similarities to the chicken-repeat-1-like elements of vertebrates. Mol Biol Evol 14:602–610
Eddy SR (1998) Profile hidden Markov models. Bioinformatics 14:755–763
Eichler EE, Clark RA, She X (2004) An assessment of the sequence gaps: unfinished business in a finished human genome. Nat Rev Genet 5:345–354
Eickbush TH, Malik HS (2002) Origins and evolution of retrotransposons. In: Craig N, Craigie R, Gellert M, Lambowitz A (eds) Mobile DNA II. ASM, Washington
Faugeron G (2000) Diversity of homology-dependent gene silencing strategies in fungi. Curr Opin Microbiol 3:144–148
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Finnegan DJ (1992) Transposable elements. Curr Opin Genet Dev 2:861–867
Fitzpatrick DA, Logue ME, Stajich JE, Butler G (2006) A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol Biol 6:99
Galagan JE, Selker EU (2004) RIP: the evolutionary cost of genome defense. Trends Genet 20:417–423
Galagan JE, Henn MR, Ma LJ, Cuomo CA, Birren B (2005) Genomics of the fungal kingdom: insights into eukaryotic biology. Genome Res 15:1620–1631
Gollotte A, L’Haridon F, Chatagnier O, Wettstein G, Arnould C, van Tuinen D, Gianinazzi-Pearson V (2006) Repetitive DNA sequences include retrotransposons in genomes of the Glomeromycota. Genetica 128:455–469
Goodwin TJ, Poulter RT (2001) The diversity of retrotransposons in the yeast Cryptococcus neoformans. Yeast 18:865–880
Goodwin TJ, Ormandy JE, Poulter RT (2001) L1-like non-LTR retrotransposons in the yeast Candida albicans. Curr Genet 39:83–91
Goyon C, Rossignol JL, Faugeron G (1996) Native DNA repeats and methylation in Ascobolus. Nucleic Acids Res 24:3348–3356
Hamer JE, Farall L, Orbach MJ, Valent B, Chumley FG (1989) Host species-specific conservation of a family of repeated DNA sequences in the genome of a fungal plant pathogen. Proc Natl Acad Sci USA 86:9981–9985
Hartl DL, Lohe AR, Lozovskaya ER (1997) Modern thoughts on an ancyent marinere: function, evolution, regulation. Annu Rev Genet 31:337–358
He C, Nourse JP, Kelemu S, Irwin JAG, Manners JM (1996) CgT1: a non-LTR retrotransposon with restricted distribution in the fungal phytopathogen Colletotrichum gloeosporioides. Mol Gen Genet 252:320–331
Heckman DS, Geiser DM, Eidell BR, Stauffer RL, Kardos NL, Hedges SB (2001) Molecular evidence for the early colonization of land by fungi and plants. Science 293:1129–1133
Hedges SB (2002) The origin and evolution of model organisms. Nat Rev Genet 3:838–849
Hibbett DS, Grimaldi D, Donoghue MJ (1997) Fossil mushrooms from Miocene and Cretaceous ambers and the evolution of Homobasidiomycetes. Am J Bot 84:981–991
Hickey DA (1982) Selfish DNA: a sexually-transmitted nuclear parasite. Genetics 101:519–531
Hood ME (2005) Repetitive DNA in the automictic fungus Microbotryum violaceum. Genetica 124:1–10
Hua-Van A, Le Rouzic A, Maisonhaute C, Capy P (2005) Abundance, distribution and dynamics of retrotransposable elements and transposons: similarities and differences. Cytogenet Genome Res 110:426–440
Johnson LJ (2007) The genome strikes back: the evolutionary importance of defense against mobile elements. Evol Biol 34:121–129
Jordan IK, McDonald JF (1999) Comparative genomics and evolutionary dynamics of Saccharomyces cerevisiae Ty elements. Genetica 107:3–13
Jordan IK, Matyunina LV, McDonald JF (1999) Evidence for the recent horizontal transfer of long terminal repeat retrotransposon. Proc Natl Acad Sci USA 96:12621–12625
Kanaya S, Oobatake M, Liu Y (1996) Thermal stability of Escherichia coli ribonuclease HI and its active site mutants in the presence and absence of the Mg2+ ion. Proposal of a novel catalytic role for Glu48. J Biol Chem 271:32729–32736
Kasuga T, White TJ, Taylor JW (2002) Estimation of nucleotide substitution rates in Eurotiomycete fungi. Mol Biol Evol 19:2318–2324
Kempken F, Kück U (1998) Transposons in filamentous fungi—facts and perspectives. Bioessays 20:652–659
Khaldi N, Collemare J, Lebrun MH, Wolfe KH (2008) Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol 9:R18
Kidwell MG (1992) Horizontal transfer. Curr Opin Genet Dev 2:868–873
Kinsey JA, Helber J (1989) Isolation of a transposable element from Neurospora crassa. Proc Natl Acad Sci USA 86:1929–1933
Kojima KK, Fujiwara H (2005) An extraordinary retrotransposon family encoding dual endonucleases. Genome Res 15:1106–1117
Kordiš D, Gubenšek F (1998) Unusual horizontal transfer of a long interspersed nuclear element between distant vertebrate classes. Proc Natl Acad Sci USA 95:10704–10709
Le Rouzic A, Capy P (2005) The first steps of transposable elements invasion: parasitic strategy vs. genetic drift. Genetics 169:1033–1043
Lovšin N, Gubenšek F, Kordiš D (2001) Evolutionary dynamics in a novel L2 clade of non-LTR retrotransposons in Deuterostomia. Mol Biol Evol 18:2213–2224
Malik HS (2005) Ribonuclease H evolution in retrotransposable elements. Cytogenet Genome Res 110:392–401
Malik HS, Eickbush TH (2000) NeSL-1, an ancient lineage of site-specific non-LTR retrotransposons from Caenorhabditis elegans. Genetics 154:193–203
Malik HS, Eickbush TH (2001) Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res 11:1187–1197
Malik HS, Burke WD, Eickbush TH (1999) The age and evolution of non-LTR retrotransposable elements. Mol Biol Evol 16:793–805
Martin SL, Bushman FD (2001) Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Mol Cell Biol 21:467–475
McClure MA, Smith C, Elton P (1996) Parameterization studies for the SAM and HMMER methods of hidden Markov model generation. Proc Int Conf Intell Syst Mol Biol 4:155–164
Medstrand P, Landry JR, Mager DL (2001) Long terminal repeats are used as alternative promoters for the endothelin B receptor and apolipoprotein C-I genes in humans. J Biol Chem 276:1896–1903
Mizrokhi LJ, Mazo AM (1990) Evidence for horizontal transmission of the mobile element jockey between distant Drosophila species. Proc Natl Acad Sci USA 87:9216–9220
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. New York, Oxford University Press
Novikova O, Sliwińska E, Fet V, Settele J, Blinov A, Woyciechowski M (2007) CR1 clade of non-LTR retrotransposons from Maculinea butterflies (Lepidoptera: Lycaenidae): evidence for recent horizontal transmission. BMC Evol Biol 7:93
Ono R, Kobayashi S, Wagatsuma H, Aisaka K, Kohda T, Kaneko-Ishino T, Ishino F (2001) A retrotransposon-derived gene, PEG10, is a novel imprinted gene located on human chromosome 7q21. Genomics 73:232–237
Peyretaillade E, Biderre C, Peyret P, Duffieux F, Méténier G, Gouy M, Michot B, Vivarès CP (1998) Microsporidian Encephalitozoon cuniculi, a unicellular eukaryote with an unusual chromosomal dispersion of ribosomal genes and a LSU rRNA reduced to the universal core. Nucleic Acids Res 26:3513–3520
Robertson HM (1993) The mariner transposable element is widespread in insects. Nature 362:241–245
Roulin A, Piegu B, Wing RA, Panaud O (2008) Evidence of multiple horizontal transfers of the long terminal repeat retrotransposon RIRE1 within the genus Oryza. Plant J 53:950–959
Sánchez-Gracia A, Maside X, Charlesworth B (2005) High rate of horizontal transfer of transposable elements in Drosophila. Trends Genet 21:200–203
Silva JC, Kidwell MG (2004) Evolution of P elements in natural populations of Drosophila willistoni and D. sturtevanti. Genetics 168:1323–1335
Slot JC, Hibbett DS (2007) Horizontal transfer of a nitrate assimilation gene cluster and ecological transitions in fungi: a phylogenetic study. PLoS ONE 2:e1097
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Taylor TN, Hass H, Kerp H (1999) The oldest fossil ascomycetes. Nature 399:648
Teng SC, Wang SX, Gabriel A (1995) A new non-LTR retrotransposon provides evidence for multiple distinct site-specific elements in Crithidia fasciculata miniexon arrays. Nucleic Acids Res 23:2929–2936
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Vivarès CP, Méténier G (2000) Towards the minimal eukaryotic parasitic genome. Curr Opin Microbiol 3:463–467
Volff JN, Körting C, Schartl M (2000) Multiple lineages of the non-LTR retrotransposon Rex1 with varying success in invading fish genomes. Mol Biol Evol 17:1673–1684
Wenzl P, Wong L, Kwang-won K, Jefferson RA (2005) A functional screen identifies lateral transfer of beta-glucuronidase (gus) from bacteria to fungi. Mol Biol Evol 22:308–316
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S et al (2002) The genome sequence of Schizosaccharomyces pombe. Nature 415:871–880
Wöstemeyer J, Kreibich A (2002) Repetitive DNA elements in fungi (Mycota): impact on genomic architecture and evolution. Curr Genet 41:189–198
Zingler N, Weichenrieder O, Schumann GG (2005) APE-type non-LTR retrotransposons: determinants involved in target site recognition. Cytogenet Genome Res 110:250–268
Župunski V, Gubenšek F, Kordiš D (2001) Evolutionary dynamics and evolutionary history in the RTE clade of non-LTR retrotransposons. Mol Biol Evol 18:1849–1863
Acknowledgements
The sequence data for P. chrysosporium, L. bicolor, T. reesei, A. niger, and P. stipitis were produced by the US Department of Energy Joint Genome Institute (http://www.jgi.doe.gov/). Preliminary sequence data for Ascosphaera apis was obtained from Baylor College of Medicine Human Genome Sequencing Center website at http://www.hgsc.bcm.tmc.edu. Preliminary sequence data for Alternaria brassicicola were obtained from Genome Sequencing Center at Washington University Medical School (http://genome.wustl.edu/index.cgi).
This work was supported in part by state contract 10002-251/П-25/155-270/200404-082 and Siberian Branch of the Russian Academy of Sciences (project No. 10.4).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
List of species, genomes of which were analyzed in silico in the present study and the sources of genomic sequences. (Word file). (DOC 88.5 KB)
Table S2
Novel non-LTR retrotransposons from fungi detected in this study and their accession numbers. (Word file). (DOC 177 KB)
Table S3
Novel non-LTR retrotransposons from fungi detected in this study, their copy number, and putative structure. (Word file). (DOC 215 KB)
Table S4
Amino acid divergences of 11 cellular proteins from A. niger, A. fumigatus, A. oryzae, F. oxysporum, and C. globosum. (Word file). (DOC 106 KB)
Fig. S1
The percentage of non-LTR retrotransposons sequences in investigated fungal genomes plotted against the genome size. (Adobe Reader file). (PDF 20.3 KB)
Fig. S2
The 50% consensus tree of the Bayesian inference based on RT amino acid sequences of non-LTR retrotransposons including newly described elements from fungi. Posterior probabilities are indicated. (Adobe Reader file). (PDF 52.9 KB)
Rights and permissions
About this article
Cite this article
Novikova, O., Fet, V. & Blinov, A. Non-LTR retrotransposons in fungi. Funct Integr Genomics 9, 27–42 (2009). https://doi.org/10.1007/s10142-008-0093-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-008-0093-8