
Abstract
Fusarium head blight (FHB), primarily caused by Fusarium graminearum Schw., is a destructive disease of wheat (Triticum aestivum L.). Although several genes related to FHB resistance have been reported, global analysis of gene expression in response to FHB infection remains to be explored. The expression patterns of transcriptomes from wheat spikes of FHB-resistant cultivar Ning 7840 and susceptible cultivar Clark were monitored during a period of 72 h after inoculation (hai) with F. graminearum. Microarray analysis, coupled with suppression subtractive hybridization technique, identified 44 significantly differentially expressed genes between cv. Ning 7840 and cv. Clark. More differentially expressed genes were identified from susceptible libraries than from resistance libraries. The up-regulation of defense-related genes in Ning 7840 relative to cultivar Clark occurred during early fungal stress (3–12 hai). Three genes, with unknown function that were up-regulated in cv. Ning 7840 at most time points investigated, might play an important role in enhancing FHB resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fusarium head blight (FHB) is an economically important disease of wheat in warm, humid, and semihumid areas worldwide. It not only significantly reduces grain yield, but also grain quality (Shroeder and Christensen 1963). Grains infected by Fusarium graminearum are often shriveled, with significantly lower kernel weight, and can be easily blown away with the chaff during threshing (Bai and Shaner 2004). Additional losses come from contamination of grains with mycotoxins produced by F. graminearum (Snijers 1990). Deoxynivalenol is a major toxin produced by the fungus during infection and is harmful to animal and human health (Desjardins and Hohn 1997). In North America, several epidemics caused by FHB in recent years have resulted in a loss of more than $1 billion per year (McMullen et al. 1997).
Currently, chemicals are not very effective for FHB control. Genetic resistance is the most effective strategy to reduce losses caused by the disease. Chinese resistant cultivar Sumai 3 and its derivative cv. Ning 7840 have been a major source of FHB resistance in most breeding programs worldwide (Bai and Shaner 2004). In cv. Ning 7840, infection is limited to the inoculated floret, and disease symptoms usually do not spread to uninoculated spikelets (Type II resistance). In contrast, disease symptoms start to spread from the point of inoculation to other spikelets of the spike of a susceptible cultivar in about 5 days after inoculation and cover the entire spike in 10 to 15 days thereafter.
Although molecular mapping of quantitative trait loci for FHB resistance has been extensively reported, studies on the biochemical and molecular bases of FHB resistance are still limited (Bai and Shaner 2004). Differences in activities of superoxide dismutase, catalase, phenylalanine ammonia-lyase, and ascorbic acid oxidase between resistant and susceptible varieties have been reported (Chen et al. 1997; Lu et al. 2001). Pritsch et al. (2000) studied the expression of peroxidase, β-glucanase, chitinase, and two thaumatin-like proteins (TLP) in spray-inoculated wheat, and reported that expression of these genes occurred as early as 6 h after inoculation (hai) with F. graminearum but was greatest at 36 to 48 hai. Results also showed that TLP were detected earlier in the resistant cv. Sumai 3 than in the susceptible cultivar Wheaton. A higher level of chitinases and β-1,3-glucanases was also observed in cv. Sumai 3 than that in its susceptible mutant during the first 24 h of pathogen infection (Li et al. 2001).
To date, limited research has been done on genes possibly involved in interaction between F. graminearum and wheat. From spray-inoculated Sumai 3 spikes sampled from 0 to 48 hai, Kruger et al. (2002) identified 29 genes related to interaction between wheat and F. graminearum. Those genes were also involved in other plant–pathogen interactions. Trail et al. (2003) identified a set of genes from the fungus that were believed to be responsible for fungal infection. Xing et al. (2000) reported three defense-related ESTs that expressed only in F. graminearum-inoculated cv. Sumai 3. These genes from the fungus and its resistant host (Sumai 3) may play an important role in the interaction between the pathogen and the host in the FHB system. More researches on this area may enlighten the mechanisms of wheat resistance to FHB infection.
DNA microarrays enable the simultaneous analysis of thousands of genes in one experiment (Freeman et al. 2000; Hedge et al. 2000), which allows global profiling of gene expression and characterization of plant defense genes responsive to biotic stresses. For instance, Puthoff et al. (2003) identified 128 genes with altered steady-state mRNA levels in response to cyst nematode parasitism in Arabidopsis. Schenk et al. (2003) observed differentially expressed Arabidopsis genes involved in plant defense, the β-oxidation pathway of fatty acids, signal transduction, and cell wall synthesis as part of the systemic acquired resistance response during the interaction between Arabidopsis and Alternaria brassicicola. However, there are no published studies on global profiling of gene expression dealing with interactions between wheat and F. graminearum during early infection stages using microarray. Genome-wide identification of up- or down-regulated genes in response to F. graminearum infection may provide a more integrated view on the responses of wheat to FHB infection. In this study, we attempted to identify differentially expressed genes between Ning 7840 (FHB-resistant) and Clark (susceptible) during early F. graminearum infection using microarray and real-time polymerase chain reaction (RT-PCR) coupled with suppression subtractive hybridization (SSH) techniques. To our knowledge, this is the first published study on global expression profiling of FHB-related genes in wheat using microarray. The information may facilitate further understanding of resistance mechanisms of wheat in response to Fusarium infection and may aid the development of an effective strategy for control of wheat FHB.
Materials and methods
Plant materials and growth conditions
Bulked segregants from a population of F9:12 recombinant inbred lines (RILs) of wheat derived from cv. Ning 7840 × cv. Clark were used to construct the SSH libraries. Cv. Ning 7840 is resistant to FHB, whereas cv. Clark is susceptible. Five resistant and five susceptible RILs were chosen to form two bulks on the basis of their FHB severity from five greenhouse tests (Bai et al. 1999). Wheat seedlings were vernalized in a germination tray for 6 weeks at 4°C in a growth chamber. After vernalization, seedlings were transplanted into 5 1/4-in. dura-pots containing Metro-Mix 360 soil (Hummert Int, Earth City, MO) and grown at 20°C for 12 h with light and at 15°C for 12 h under darkness. For the microarray analysis, Ning 7840 and Clark were grown under the conditions described above.
Inoculation of wheat spike and mRNA isolation
F. graminearum conidiospores were produced as described by Bai et al. (2000). At anthesis, 10 μl of F. graminearum conidiospore suspension (100 spores/μl) was injected into a central floret of a spike with a pipette. The inoculated spikes were enclosed with sandwich bags misted with water inside until sampling. All inoculated plants were grown in a growth chamber at 25°C for 18 h of light and at 22°C for 6 h of darkness. After removal of the inoculated floret, the rest of the wheat spike was harvested, immediately frozen in liquid N2, and stored at −80°C until use.
SSH libraries
Suppression subtractive hybridization (SSH) was done using the PCR-select cDNA subtraction kit from Clontech (Palo Alto, CA) following manufacturer’s instructions. Messenger RNA was extracted from tissues of bulked susceptible or resistant lines (0.5 g/line) with the Message Maker Kit (Invitrogen, Carlsbad, CA). Tissues collected at 6, 36, and 72 hai were used to generate SSH libraries. The forward libraries used mRNA from the inoculated resistant RILs as the tester and the inoculated susceptible RILs as the driver and vice versa for the reverse library. PCR products from SSH were cloned into a pGEM-T Easy vector (Promega, Madison, WI).
Microarray analysis
Complementary DNA inserts in cloning vector were amplified via PCR in a 100-μl total reaction volume consisting of 4 μl of fresh bacterial cells containing target PCR fragments, 1× PCR buffer, 1.5 mM MgCl2, 0.24 mM dNTPs, 0.26 μM each of M13 primers, and 2 U of Taq polymerase (Promega). All PCR products were analyzed in a 1% agarose (w/vol) gel in 1× TAE buffer to assess the quality of amplicons. Only the clones that produced bright and sharp PCR products were printed in the array. The PCR products (total of 2,306) were precipitated with 3 M NaOAC (pH 5.2) and two volumes of ethanol. The resulting pellet was resuspended in 3× SSC at a final concentration of 100 ng/μl. The PCR products, actin gene (positive control), and vector with no insert, 3× SSC, and water (negative controls) were arrayed on GAPS II coated slides (Corning, Corning, NY) by using an OmniGrid100 Microarray Printer (GeneMachines, San Carlos, CA) at the Microarray Core Facility, Oklahoma State University (Stillwater, OK). A total of 4,806 cDNA clones were arrayed on a glass slide. Among them, 2,306 clones were isolated from six SSH libraries that derived from the inoculated spikes after 6, 36, and 72 hai. In addition, 2,500 clones from aluminum-stressed (Al) wheat libraries, derived from near-isogenic lines contrasting in Al tolerance (Xiao et al. 2005; Guo et al. personal communication), were also spotted on the same slide, so the arrays can be used for both studies on FHB and Al-stress. In brief, Chisholm-T (Al tolerant) and Chisholm-S (Al sensitive), which were subjected to 6 h and 1 day stress with 10 mg/l Al3+, were used to construct SSH cDNA libraries by using PCR-select cDNA subtraction kit (Clontech) according to the manufacturer’s instruction. Forward subtraction used mRNA from Chisholm-T as a tester and Chisholm-S as a driver and vice versa for the reverse subtraction.
Samples had three replicates and printed at the same interval for technical replication in each slide. Complimentary DNAs were immobilized in the glass slides by cross-linking at 300 mJ with a Stratalinker (Stratagene, La Jolla, CA). Before hybridization, the slide was placed over a beaker with boiling water for a few seconds with the array side facing down to rehydrate the array. Once a thin layer of mist covered the slide surface, the slide was removed and dried briefly by placing the slide face up on a hot plate (30 s) then baking in an 80°C oven for 3 h. Slides were stored desiccated in vacuo at room temperature until use.
F. graminearum-inoculated spikes of cv. Ning 7840 and cv. Clark were sampled at 0, 3, 6, 12, 24, 36, 48, and 72 hai. Two wheat spikes per time point were pooled for RNA extraction using Trizol reagent (Invitrogen). 3DNA Array 50 kit from Genisphere (Genisphere, Hatfield, PA) was used to detect differentially expressed genes between fungal-inoculated FHB resistant and susceptible cultivars. Microarray probes were synthesized from equal amounts of total RNA isolated from the spike of Ning 7840 and Clark at different time points. Complimentary DNA hybridizations were conducted overnight at 45°C in a Corning hybridization chamber (Corning). After washing, 3DNA hybridization was performed at 48°C for 3 h to incorporate Cy5 or Cy3 fluorochrome. Hybridizations were repeated twice for all time points of the fungal stress, and dyes were swapped to avoid bias in dye intensity detection.
Data analysis
The signal intensity in each array was captured by scanning the slides at two wavelengths (532 nm for Cy3 and 635 nm for Cy5) using GenePix 4000B microarray scanner (Axon Instruments, Union City, CA) at pixel size resolution of 10 microns. The Cy5 and Cy3 images were combined and spot fluorescence intensity was analyzed using GenePix Pro 5.0 software (Axon). The composite image was visually inspected and the spots with scratches or dust particles were excluded from further analysis. In addition, any sample with signal intensity less than 200 in both channels or outliers (spots with signal intensity greater or less than the mean log2 ratio +2 SDs among replicates) were removed. The signal intensity values of the actin housekeeping genes were used to normalize the data. The GenePix Pro output was statistically analyzed for differential gene expression between Ning 7840 and Clark using R-project statistical environment (http://www.r-project.org) and Bioconductor (http://www.bioconductor.org) through the GenePix AutoProcessor (http://www.darwin.biochem.okstate.edu/). The log2 ratios of replicated spots were averaged, and differentially expressed genes were ranked from the highest to the lowest based on significance of B statistics. Differential expression of a gene was considered to be significant if a B value is at least 5 (P<0.01). Genes with similar expression patterns were grouped together by using K-means clustering module of Genesis software (Sturn et al. 2003).
Gene sequence analysis
Bacteria with cloned EST fragments were grown in 96-well plates with Circle growth medium (Bio 101, San Diego, CA) for 20 h at 37°C on a shaker at 200 rpm. DNA was extracted using a QiaPrep Turbo 96 BioRobot Kit (Qiagen, Valencia, CA) and sequenced with M13 universal primers. Sequencing was run either in a 3700 ABI automated sequencer or in a Li-Cor 4200 DNA analyzer (Li-Cor, Lincoln, NE) with a DYEnamic Direct Cycle Sequencing Kit (Amersham Biosciences, Piscataway, NJ). A BLAST-X sequence homology search (Altschul et al. 1997) was performed against the nonredundant protein database in GenBank. The highest similarity score was considered as the best match for the putative identity of corresponding ESTs. Sequence similarity was considered significant if E-value was equal to or less than 0.001. MIPS Functional Catalogue (http://www.mips.gsf.de/proj/funcatDB/) was used to classify ESTs.
Real-time polymerase chain reaction
The same batch of RNA samples from 3, 12, 36, and 72 hai that was initially extracted for the microarray experiment was used for real-time polymerase chain reaction (RT-PCR). First-strand cDNA synthesis was performed in a 25-μl reaction by combining 10 μg total RNA with 1 pmol oligo(dT)12-18 (Invitrogen) and random primers (Promega). The mix was incubated at 80°C for 10 min before transferring to an ice tub. Twenty units of SUPERaseIn Rnase Inhibitor (Ambion, Austin, TX) was added to the RNA-primer mix. A 50_μl final reaction consisting of 10 μl 5X Superscript II first strand buffer, 3 μl of 10 mM dNTPs, 5 μl 0.1 M dithiotreitol, 200 U Superscript II enzyme (Invitrogen), and the RNA-primer mix was incubated at 42°C for 2 h.
Quantitative PCR reactions (25 μl total volume) were carried out in 96-well UV plates containing iQ Sybr Green Supermix (Bio-Rad Laboratories, Hercules, CA), 10 pmol each of forward and reverse gene-specific primers (Supplemental Material Table S1), and 10.5 μl cDNA sample (1:1,000 dilution). Gene-specific primers were designed using Beacon Designer software (Premier Biosoft International, Palo Alto, CA). PCR amplification was performed in an iCycler (Bio-Rad Laboratories, Hercules, CA) starting with a denaturation step at 95°C for 5 min, followed by 45 cycles of 95°C for 20 s and 60°C for 1 min. After amplification, a melting step was performed at 95°C for 1 min, then cooling down to 55°C for 1 min, followed by a slow rise in temperature to 95°C at a rate of 0.5°C/10 sec. Melt-curve analysis was performed after each RT-PCR run to verify the specificity of Sybr Green dye and absence of primer-dimer. To normalize the total amount of cDNAs present in each reaction, wheat β-actin housekeeping gene was co-amplified. Time points of 3, 12, 36, and 72 hai were assayed for each gene. Each sample was replicated twice, and the ΔΔCT method of relative gene quantification (Applied Biosystems 1997) was used to calculate the expression level of Ning 7840 relative to Clark.
Results
SSH library and microarray analysis
A total of 44 significantly differentially expressed genes were identified between cv. Ning 7840 and cv. Clark (Table 1). Among them were 29 genes from FHB-stress libraries and 15 genes from Al-stress libraries. DNA sequence homology search identified 20% of the genes as defense-related and 21% to be involved in metabolism, transport facilitation, transcription regulation, and signal transduction. Majority of the genes have unknown function (23%) or no homology with known accessions in the GenBank (36%). All genes that produced a significant BLAST hit were similar to plant proteins. Fungal protein was not identified.
Expression profiles of differentially expressed genes
The differentially expressed genes were grouped into six clusters using K-means method (Fig. 1). On the basis of their expression patterns, these clusters were further reclassified into four major groups: (1) up-regulated in Ning 7840 in comparison to Clark at most time points investigated, (2) up-regulated in Ning 7840 mainly at early time points and down-regulated at later time points of fungal stress, (3) down-regulated in Ning 7840 at later time points of fungal stress, and (4) down-regulated in Ning 7840 during all time points studied. Among the 44 differentially expressed genes, seven have two to five identical copies each (data not shown). The redundant genes had the same expression profile and mapped to the same cluster; therefore, only one representative was reported hereafter. These results also demonstrate good repeatability of our microarray experiment.

Mean expression level of six clusters of differentially expressed genes between Ning 7840 and Clark after fungal stress. The genes were grouped based on similarity of expression pattern (K-means) using Genesis software. The x-axis represents the time point at which spikes were sampled, while the y-axis shows differential expression in log-based two ratios. The lines represent the mean transcript abundance at the time course of infection and the bars represent ±SD. A log2 ratio of “+1” and “+2” mean that the expression of a gene in Ning 7840 is up-regulated by two-and fourfold, respectively, relative to Clark; whereas “−1” and “−2” represent down-regulation of gene expression in Ning 7840 by two- and four-fold, respectively, compared to Clark
Cluster b and e belong to group 1. All three genes in cluster b showed significant up-regulation in most time points of the fungal stress in Ning 7840 (Fig. 1b). One gene in this group encoded a kinesin, heavy, chainlike protein, and two had similarity with unknown genes in GenBank (Table 1). Cluster e contains three genes that were all strongly up-regulated in the resistant cv. Ning 7840 at all time points of the fungal stress, with expression peak at 6 hai (Fig. 1e). All genes in this cluster were from the library of Ning 7840 after 6 h of inoculation. Two of them are similar to a putative transposable element and serine protease, while the other is similar to an unknown protein.
Group 2 is the largest group (18 genes) and consists of clusters a and f. Genes in this group were up-regulated in Ning 7840 before or during early infection and then down-regulated (Fig. 1a,f). Genes in this cluster are mainly involved in cell rescue and defense (Table 1). Most of the genes in this group were up-regulated in cv. Ning 7840 at 0 and 3 hai. However, the expression of these genes gradually decreased in Ning 7840, and became significantly down-regulated at 24 to 72 hai. Genes in this group have significant similarity with defense-related genes such as pathogenesis-related (PR) proteins, P450s, potassium transporter and proteinase/protease inhibitors. Nine genes in this group have unknown function.
Cluster d belongs to Group 3. This cluster includes seven genes: calreticulin, P450, two PR proteins, NPR-1, 3-beta hydroxysteroid dehydrogenase/isomerase, and PDR-like ABC transporter (Table 1). The putative functions of the other nine genes are unknown. Genes in this cluster were significantly down-regulated in Ning 7840 at 36 to 72 hai (Fig. 1d). Most genes did not show differential expression for other time points, but genes such as the PR protein and three unknown genes (C124, G8-345, and G8-34) were significantly up-regulated at 6–12 hai (data not shown).
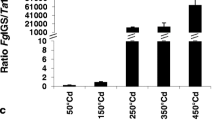
Group 4 contains four genes from cluster c (Table 1). Genes from this group were mainly down-regulated at all time points tested (Fig. 1c). Most of them have either no significant similarity with any gene in GenBank or similarity to the genes without known function. The gene with unknown function in this group is a PR protein.
Validation of differentially expressed genes by RT-PCR
Changes in expression of eight genes identified in the microarray experiment were verified using quantitative RT-PCR. RT-PCR data for each gene was normalized based on expression of a wheat housekeeping gene β-actin. The expression of β-actin was uniform across all treatments (Fig. S1). In most cases, a much larger increase in fold expression was obtained from RT-PCR than that from microarray analysis (Fig. S2). Microarrays have a relatively low dynamic range due to low concentrations of cDNA deposited in slides (Ozturk et al. 2002). As such, they sometimes do not reflect the full changes in transcript abundance especially for those strongly up- or down-regulated transcripts. However, the trend of up- or down-regulation of a gene in Ning 7840 was consistent (Fig. S2). The result indicated a good agreement between the results from microarray and RT-PCR.
Discussion
Suppression subtractive hybridization
SSH was the technique of choice for library construction because it involves a normalization step to balance the abundance of cDNA in the target population and a subtraction step to remove cDNA that are common between the target and tester populations (Diatchenko et al. 1996). Another advantage of SSH is that it can enrich rare cDNA (several molecules per cell) by more than 1,000-fold (Diatchenko et al. 1996), thereby enabling the isolation of rare transcripts that may not be easily obtained from a regular cDNA library. Any gene that is commonly induced in two populations as a response to fungal stress will theoretically be subtracted out. In this study, a very small subset of genes (2,306) were derived from six FHB-stress SSH libraries instead of a library of all genes from whole-wheat genomes, which made it feasible for us to focus on a small number of genes with limited resources. Therefore, SSH greatly reduced the number of clones to be screened to a more manageable size, significantly reducing screening work and cost. In addition, the SSH libraries were constructed by using two bulks of recombinant inbred lines with extreme responses to FHB infection. Pooled samples are expected to average out the contribution of background genes that is not related to FHB resistance or susceptibility.
Previous researches indicated that the Fusarium macroconidia usually germinate later than 6 hai, enter the floret tissue by 36 hai, and spread to nearby uninoculated spikelets after 48 hai (Pritsch et al. 2000, 2001; Kang and Buchenauer 2000). In this study, SSH libraries were constructed with samples collected at 6, 36, and 72 hai that covered an important time period of early fungal infection from spore germination to spread of symptoms to uninoculated spikelets.
Defense-related genes are up-regulated during early stages of fungal stress
Coordinately regulated genes in response to F. graminearum infection were identified by cluster analysis in this study. Group 3, with genes from clusters a and f, contains putative genes for plant defense that were up-regulated in Ning 7840 during the early stages of fungal stress (0 to 3 hai). Among them, a chitinase II precursor and PR-1 gene showed up-regulation in cv. Ning 7840 at early time points of fungal stress, then down-regulated from 24 hai onward. Chitinase can degrade chitin in fungal cell walls (Collinge et al. 1993) and releases oligomeric products that can signal a plant to activate additional defense responses (Li et al. 2001). The chitinase precursor identified in this study is similar to the wheat chitinase reported by Li et al (2001). Similar expression patterns were observed for these two chitinase genes wherein their expression peaks occurred in FHB-inoculated resistant genotypes at 12 hai or earlier, and a higher level of gene expression was observed in the susceptible genotypes at 48 hai (Li et al. 2001). Abundant PR-1 and chitinase genes were also found in a F. graminearum-infected Sumai 3 library (Kruger et al. 2002). Another PR gene, up-regulated at 6 to12 hai and down-regulated at 48 to 72 hai in this study, was similar to a jasmonate-inducible PR-10 gene (Table 1, cluster d). PR-10 can also be induced by the fungal pathogen Magnaporthe grisea in rice (Jwa et al. 2001). Differential expression of the genes for chitinase and PR-proteins may play important roles in activation of early defense responses in resistant genotypes.
Three genes in cluster a that share significant homologies with putative P450 genes were up-regulated at early time points of fungal stress. One P450 gene (6F88) belongs to the CYP71C subfamily and may participate in the biosynthesis of 2,4-dihydroxy-7-methoxy-1,4-benzoxazin-3-one (DIMBOA), a major antifungal compound in maize, wheat, and rye (Frey et al. 1997). DIMBOA inhibits proteases and oxidative enzymes in fungi, bacteria, and insects (Niemeyer 1988). DIMBOA-derived antimicrobial compounds, 6-methoxy-2-benzoxazolinone and 2-benzoxazolinone, significantly inhibited the growth of F. graminearum in vitro (Glen et al. 2001). The P450 gene identified in this study may hinder the growth of F. graminearum and development of FHB symptoms. The function of the other two P450s is still unknown.
Two genes in cluster f are protease/proteinase inhibitors that were up-regulated in Ning 7840 at 3 hai. Dunaevskii et al. (1997) showed that the proteinase inhibitor trypsin/chymotrypsin inhibited germination and growth of Alternaria alternata in buckwheat. Cordero et al. (1994) demonstrated the up-regulation of a proteinase inhibitor gene in corn after the plant was wounded or infected by fungal pathogens. This enzyme can also disrupt the protein metabolism of insects that attack a plant after the plant is wounded (Hammond-Kosack and Jones 1996). The up-regulation of proteinase inhibitors at 3 hai in cv. Ning 7840 may play a role in inhibiting germination of F. graminearum in Ning 7840.
A putative potassium transporter was uniquely expressed during Sumai 3–F. graminearum interaction (Kruger et al. 2002). A similar transporter gene was up-regulated in Ning 7840 during early fungal infection and, therefore, may be involved in defense against infection of F. graminearum in a wheat spike.
In this study, the expression of PR proteins, proteinase inhibitor-like protein, and cytochrome P450 were down-regulated in Ning 7840 with respect to Clark at 36 to 72 hai. Down-regulation in Ning 7840 means that expression of these genes is significantly higher in the susceptible cv. Clark during late time points of infection initiation. Drought stress also induced a high level of expression of the similar genes in barley (Ozturk et al. 2002). In F. graminearum-infected Clark, drought stress probably occurs after fungal spread to uninoculated spikelets after 48 hai (Pritsch et al. 2000). Pathogen spread causes clogging of the rachis, thereby limiting or blocking nutrient and water supply to spikelets above the infected floret. This suggests that fungal spread within a spike in a susceptible genotype may induce the expression of drought-stress genes.
Other differentially expressed genes
A group of three genes in cluster e was consistently up-regulated in the resistant cv. Ning 7840. All three ESTs originated from the libraries of cv. Ning 7840 (forward). Gene 6F89 had a similarity with a putative transposable element, 6F19 with a serine protease, whereas the function of the other gene is still unknown. As these genes were always expressed to a greater extent in the resistant cultivar than in the susceptible cultivar at most time points in this study, these could be very important genes for FHB resistance. Further investigation of the functions of these genes may provide useful information for the understanding of the mechanisms of FHB resistance.
On the other hand, all but one of the differentially expressed genes derived from FHB-induced library in clusters c and d were from FHB-susceptible libraries. Genes in the two clusters were down-regulated in Ning 7840 either at all time points or at later time points of fungal stress. Therefore, the expression level of these genes in cv. Clark was significantly higher than that in cv. Ning 7840. One of the down-regulated genes in this cluster had significant similarity with a putative NPR1 regulatory gene. This gene was reported as a necessary gene for the activation of PR genes in Arabidopsis, and overexpression of this gene resulted in more resistant Arabidopsis plants (Cao et al. 1998). However, in wheat, overexpression of wheat NPR1 resulted in increased susceptibility (Rommens and Kishore 2000). Further investigation of these genes may elucidate their role in wheat-susceptible response to FHB infection.
In this study, many of the differentially expressed genes (86%) came from the susceptible libraries, and only 14% were from libraries derived from Ning 7840. This observation may indicate that more transcripts were expressed at higher levels in Clark as a response to F. graminearum infection. Microarray analyses using cv. Ning 7840 inoculated with F. graminearum vs inoculated with mung bean medium showed only 25% as many significantly differentially expressed genes as those in Clark (unpublished data). The small number of genes derived from cv. Ning 7840 could be due to (1) some genes involved in FHB resistance of cv. Ning 7840 are not regulated at transcription level or (2) disease resistance in cv. Ning 7840 may result from a loss of function of susceptibility genes. Further investigation of the roles of the susceptibility genes in FHB infection process may provide insight into the understanding of the resistance mechanism of FHB in wheat.
The identification of many differentially expressed genes between cv. Ning 7840 and cv. Clark suggests that plant defense in response to FHB infection is a complex regulatory network of genes involved in signal transduction, metabolism, transport facilitation, and cell rescue/defense, as well as genes of still unknown function. Although a large percentage of these genes produced no hit in public databases or were similar to hypothetical or unknown proteins, similarities in gene expression pattern with known genes provide possible insight into the functions for novel or unknown proteins. Further characterization of these genes may help to understand the molecular basis of FHB resistance and lay a solid ground for cloning of FHB resistance genes in wheat.
References
Applied Biosystems (1997) Relative quantification of gene expression. ABI PRISM 7700 sequence detection system user bulletin #2, pp 11–15
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bai GH, Kolb FL, Shaner GE, Domier LL (1999) Amplified fragment length polymorphism markers linked to a major quantitative trait locus controlling scab resistance in wheat. Phytopathology 89:343–348
Bai GH, Shaner GE (2004) Management and resistance in wheat and barley to Fusarium head blight. Annu Rev Phytopathol 42:135–161
Bai GH, Shaner G, Ohm H (2000) Inheritance of resistance to Fusarium graminearum in wheat. Theor Appl Genet 100:1–8
Cao H, Li X, Dong X (1998) Generation of broad-spectrum disease resistance by overexpression of an essential regulatory gene in systemic acquired resistance. Proc Natl Acad Sci U S A 95:6531–6536
Chen LF, Song Y, Xu YG, Nie L, Xu LL (1997) Comparison of activities of superoxide dismutase and catalase between scab-resistant and susceptible wheat varieties. Acta Phytopathologica Sinica 27(3):209–213
Collinge DB, Kragh KM, Mikkelsen JD, Nielsen KK, Rasmussen U, Vad K (1993) Plant chitinases. Plant J 3:31–40
Cordero MJ, Raventos D, San-segundo B (1994) Expression of a maize proteinase inhibitor gene is induced in response to wounding and fungal infection: systemic wound response of a monocot gene. Plant J 6:141–150
Desjardins AE, Hohn TM (1997) Mycotoxins in plant pathogenesis. Mol Plant-Microb Interact 10:147–152
Diatchenko L, Lau YFC, Campbell PA, Chenchik A, Moqadom F, Huang B, Lukyanov K, Gurskaya N, Sverdlov ED (1996) Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci U S A 93:6025–6030
Dunaevskii YE, Gladysheva IP, Pavlukova EB, Beliakova GA, Gladyschev DP, Papisova AI, Larionova NI, Belozersky MA (1997) The anionic protease inhibitor BBWI- 1 from buckwheat seeds: kinetic properties and possible biological role. Physiol Plant 100:483–488
Freeman WM, Robertson, DJ Vrana KE (2000) Fundamentals of DNA hybridization arrays for gene expression analysis. BioTechniques 29:1042–1055
Frey M, Chomet P, Glawischnig E, Stettner C, Grün S (1997) Analysis of a chemical plant defense mechanism in grasses. Science 277:696–699
Glen AE, Hinton DM, Yates IE, Bacon CW (2001) Detoxification of corn antimicrobial compounds as the basis for isolating Fusarium verticillioides and some other Fusarium species from corn. Appl Environ Microbiol 67:2973–2981
Hammond-Kosack KE, Jones JDG (1996) Resistance gene-dependent plant defense responses. Plant Cell 8:1773–1791
Hedge P, Qi R, Abernathy K, Gay C, Dharap S, Gaspard R, Hughes JE, Snesrud E, Lee N, Quackenbush J (2000) A concise guide to cDNA microarray analysis. BioTechniques 29:548–562
Jwa N-S, Agrawal GK, Rakwal R, Park C-H, Agrawal VP (2001) Molecular cloning and characterization of a novel jasmonate-inducuble pathogenesis-realted class 10 protein gene, JIOsPR10, from rice (Oryza sativa L.) seedling leaves. Biochem Biophys Res Commun 286:973–983
Kang Z, Buchenauer H (2000) Ultrastructure and immunocytochemical investigation of pathogen development and host responses in resistant and susceptible wheat spikes infected by F. culmorum. Physiol Mol Plant Pathol 57:255–268
Kruger WM, Pritch C, Staggs R, Muehlbauer GJ (2002) Functional and comparative bioinformatic analysis of expressed genes from wheat spikes infected with Fusarium graminearum. Mol Plant-Microb Interact 15:445–455
Li WL, Faris JD, Muthukrishnan S, Liu DJ, Chen P, Gill BS (2001) Isolation and characterization of novel cDNA clones of acidic chitinases and β-1, 3-glucanases from wheat spikes infected with Fusarium graminearum. Theor Appl Genet 102:353–362
Lu WZ, Chen SH, Wang YZ (eds.) (2001) Research on wheat scab. Beijing, China: Sci Publ House, pp. 136–172
McMullen M, Jones R, Gallenberg D (1997) Scab of wheat and barley: a re-emerging disease of devastating impact. Plant Dis 81:1340–1348
Niemeyer HM (1988) Hydroxamic acids (4-hydroxy-1,4-benzoxazin-3-ones): defence chemicals in the Gramineae. Phytochemistry 27:3349–3358
Ozturk ZN, Talame V, Deyholos M, Michalowski CB, Galbaith DW, Gozukirmisi N, Tuberosa R Bonert HJ (2002) Monitoring large-scale changes in transcript abundance in drought and salt-stressed barley. Plant Mol Biol 48:551–573
Puthoff DP, Nettleton D, Rodermel SR, Baum TJ (2003) Arabidopsis gene expression changes during cyst nematode parasitism revealed by statistical analyses of microarray expression profiles. Plant J 33:911–921
Pritsch C, Muehlbauer GJ, Bushnel WR, Somers DA, Vance CP (2000) Fungal development and induction of defense response genes during early infection of wheat spikes by Fusarium graminearum infection. Mol Plant-Microb Interact 13:159–169
Pritsch C, Vance CP, Bushnel WR, Somers DA, Hohn T.M, Muehlbauer GJ (2001) Systemic expression of defense response genes in wheat spikes as a response to Fusarium graminearum infection. Physiol Mol Plant Pathol 58:1–12
Rommens CM, Kishore GM (2000) Exploiting the full potential of disease-resistance genes for agricultural use. Curr Opin Biotechnol 11:120–125
Schenk PM, Kazan K, Manners JM, Anderson JP, Simpson RS, Wilson IW, Somerville SC, MacLean DJ (2003) Systemic gene expression in Arabidopsis during an incompatible interaction with Alternaria brassicicola. Plant Physiol 132:999–1010
Shroeder HW, Christensen JJ (1963) Factors affecting resistance of wheat to scab caused by Giberella zeae. Phytopathology 53:831–838
Snijers CHA (1990) Fusarium head blight and mycotoxin contamination of wheat, a review. Neth J Plant Pathol 96:187–198
Sturn A, Mlecnik B, Pieler R, Rainer J, Truskaller T, Trajanoski Z (2003) Client-server environment for high-performance gene expression data analysis. Bioinformatics 19:772–773
Trail F, Xu J-R, San Miguel P, Halgren RG and Kistler HC (2003) Analysis of expressed sequence tags from Gibberella zeae (anamorph Fusarium graminearum). Fungal Genet Biol 38:187–197
Xiao K, Bai G, Carver B (2005) Nylon filter arrays reveal differential expression of ESTs in wheat roots under aluminum stress. J Integr Biol 47(7):839–848
Xing DH, Yen Y, Rudd JC, Jin Y (2000) Identification, cloning and sequencing of ESTs related to FHB resistance of wheat. In: 2000 National Fusarium Head Blight Forum, Erlanger, KY, 10–12 Dec, pp. 62–63
Acknowledgements
This work was partially supported by the Great Plains Biotechnology Consortium and the US Wheat and Barley Scab Initiative. We thank Dr. Anne Desjardin for providing the GBZ 3639 strain of F. graminearum; the Oklahoma State University Microarray Core Facility for providing access to microarray equipment, which was supported by the grants from NSF (EOS-0132534) and NIH (2P20RR016478-04, 1P20RR16478-02, and 5P20RR15564-03); and Dr. Jianfa Bai at the Gene Expression Facility at Kansas State University for technical support on RT-PCR. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the US Department of Agriculture. This is contribution No. 05-277-5 from the Kansas Agricultural Experiment Station, Manhattan, KS.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Table S1
Sequence of primers used for real time PCR (PDF 23 kb)
Figure S1
Fluorescence signal of eight cDNA templates amplified with wheat beta-actin primers (non-regulated control) indicate equal template concentrations (PDF 45kb)
Figure S2
Microarray (A) and real-time PCR (R) data in log2 ratios from a selected set of genes representing 6 clusters. Log2 ratios are on the Y-axis while the X-axis denotes the time point of inoculation. Letters to the right of the each graph indicate the gene cluster. A positive value indicates up-regulation of gene expression and a negative value indicates down-regulation of gene expression in resistant cv. Ning7840 relative to susceptible cv. Clark. RT-PCR values are from an average of two replicates (PDF 45 kb)
Rights and permissions
About this article
Cite this article
Bernardo, A., Bai, G., Guo, P. et al. Fusarium graminearum-induced changes in gene expression between Fusarium head blight-resistant and susceptible wheat cultivars. Funct Integr Genomics 7, 69–77 (2007). https://doi.org/10.1007/s10142-006-0028-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-006-0028-1