
Abstract
The bacterium Staphylococcus aureus is a common cause of human infection, and it is becoming increasingly virulent and resistant to antibiotics. Our understanding of the evolution of this species has been greatly enhanced by the recent sequencing of the genomes of seven strains of S. aureus. Comparative genomic analysis allows us to identify variation in the chromosomes and understand the mechanisms by which this versatile bacterium has accumulated diversity within its genome structure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Bacterial genetics and evolution
The increasing levels of antibiotic resistance and the emergence of epidemic strains of bacterial pathogens over the last decade (Enright et al. 2002; Livermore 2003) highlight the adaptability of bacteria and the remarkable speed of bacterial evolution. In the face of constant environmental challenges, the ability of bacteria to generate genetic variation is crucial for their survival. Bacterial genomes tend to evolve through several routes: mutation to existing genes, DNA loss or rearrangement or horizontal transfer of gene/s from one bacterium to another (Ziebuhr et al. 1999). Selective pressures exerted by environmental conditions promote the survival of cells that possess advantageous genes (Preston et al. 1998). The haploid nature of a bacterial genome means that there is a direct link between allele and phenotype. The speed of bacterial growth and division, coupled with their asexual reproduction, ensures that a successful allele can become prevalent in a clonal population very quickly. Adaptation via mutation tends to be a slow process, whereas horizontal gene transfer allows a bacterium to adapt to a new niche very rapidly (Hacker and Carniel 2001).
Recent years have seen a bonanza of bacterial genomes, with the publication of more than 200 complete genomes (for a comprehensive list of the publicly available genomes, see http://www.ebi.ac.uk/genomes/bacteria.html). For several species, there are now sequences for more than one strain available (Bolotin et al. 2004; Cazalet et al. 2004; Fouts et al. 2005; Tettelin et al. 2005). As a result, we are now able to examine genetic variation on a genomic scale and map the evolutionary changes that define closely related bacteria. This has proved to be a very valuable approach in understanding the mechanisms of evolution of bacterial pathogens. In this review, we are going to focus on the versatile pathogen Staphylococcus aureus. Six genome sequences have been published for this organism (Baba et al. 2002; Gill et al. 2005; Holden et al. 2004; Kuroda et al. 2001). By focusing on the differences in the genomes, we aim to illustrate the mechanisms that generate change in S. aureus and how these shape its virulence and drug resistance.
S. aureus as a pathogen
S. aureus is a bacterium that has gained notoriety over recent years due to its ability to evolve new virulent and drug-resistant variants (Chambers 2005; Pearson 2002). In particular, the spread of S. aureus in hospitals has placed an increased burden on health care systems (Kim et al. 2001; Nathwani 2003). S. aureus is the most common cause of hospital-acquired infection and is estimated to cause clinical disease in 2% of all patient admissions (Emmerson et al. 1996; Jones 2003). Accompanying the spread of this bacterium has been an increase in the resistance to antibiotics. In parts of Europe, the United States and Japan, 40–60% of all hospital S. aureus infections are now resistant to the β-lactam antibiotic methicillin (Fluit et al. 2001; Tiemersma et al. 2004). Methicillin-resistant S. aureus (MRSA) strains were first described in the 1960s; however, MRSAs did not spread rapidly until the late 1980s to mid-1990s, such that they are now endemic in hospitals and cannot be eradicated (Enright et al. 2002). MRSA infections are treated with vancomycin, but the first high-level vancomycin-resistant isolates (VRSA) were recently described (Anonymous 2002a,b; Chang et al. 2003). There are few reliable antibiotics on the market for treating MRSA infections, and fewer are expected to be developed in the near future (Projan and Shlaes 2004).
S. aureus is a persistent resident of the human nose in 20% of the population and intermittently carried by another 60% (Kluytmans et al. 1997). For most healthy individuals, colonization is not a problem; however, if S. aureus contaminates a breach in the skin or mucous membranes, it can go on to infect any tissue of the body. Patients in hospital are often particularly prone to S. aureus infection due to compromised immune systems and frequent catheter insertions and injections. Infections range from the trivial, such as minor skin infections, including boils and carbuncles, to more serious and life-threatening conditions, such bacteremia, endocarditis and haemolytic pneumonia. Hospitalized patients with S. aureus bacteremia have a mortality rate of at least 20%, and those with endocarditis and haemolytic pneumonia have a mortality rate of more than 20% (Cosgrove et al. 2003).
Whilst the S. aureus infection rates are lower outside the hospital, there is an emerging problem of community-acquired severe invasive infections caused by S. aureus (Chambers 2005). In recent years, increasingly virulent strains of S. aureus have emerged in the community, causing severe skin and soft tissue infections and a lethal form of haemolytic pneumonia in children (Dufour et al. 2002a; Gillet et al. 2002; Herold et al. 1998). These new strains are associated with a toxin called Panton–Valentine leucocidin, and some are also resistant to methicillin and flucloxacillin, the first-line antibiotics for community infections. In comparison to hospital-acquired strains, the drug resistance of community-acquired strains tends to be lower. However, the last decade has seen an increase in the number of community-acquired MRSA strains reported (Zetola et al. 2005).
S. aureus genome sequencing
Currently, there are six published S. aureus genome sequences from strains isolated from different clinical settings and with varied antibiotic resistances: N315 and Mu50 are hospital-acquired MRSA strains, the latter of which has decreased susceptibility to the antibiotic vancomycin (VISA) (Kuroda et al. 2001); MW2 is a community-acquired MRSA (Baba et al. 2002); MRSA252 is a hospital-acquired MRSA strain (Holden et al. 2004) and representative of the highly successful epidemic EMRSA-16 clonal group responsible for 50% of UK MRSA infections and one of the major MRSA clones found in the USA (USA200) (Johnson et al. 2001; McDougal et al. 2003); MSSA476 is a community-acquired methicillin-susceptible strain (Holden et al. 2004); and COL is an early MRSA strain originally isolated in the 1960s (Gill et al. 2005). In addition, the complete genome of NCTC8325, a genetically tractable laboratory strain, is also freely available (http://www.genome.ou.edu/staph.html).
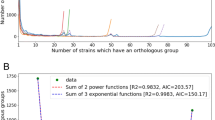
The sequenced S. aureus genomes range in size from 2.813 to 2.903 Mb and comprise a single circular chromosome, and in some cases a plasmid, and are predicted to contain between 2,592 and 2,748 genes. The overall structure of the S. aureus chromosomes is well conserved, with the majority of genes exhibiting high levels of DNA identity (>97%) and organized in the same order, interrupted by small regions of difference (Fig. 1). If the comparison is widened to include related bacteria from the same genus (Staphylococcus epiderimidis) (Gill et al. 2005) and order (Bacillus subtilis) (Kunst et al. 1997), the level of gene order conservation falls away with the taxonomic diversity (Fig. 2a). However, the detectable blocks of gene conservation in the most distant comparison represent the conservation of chromosomal structure maintained over millions of years since these bacteria diverged from a common ancestor (Battistuzzi et al. 2004). Many of these regions are concerned with central metabolism and housekeeping functions. Taxonomic diversity is also reflected in the number of orthologous genes, i.e. those genes that are derived from a common ancestor and whose products are predicted to perform the same function or role; the closer the relatedness, the higher the number of orthologues (Fig. 2b).

Comparison of the chromosomes of six sequenced Staphylococcus aureus strains. Pairwise comparisons of the S. aureus COL, Mu50, N315, MW2, MSSA476 and MRSA252 chromosomes displayed using the Artemis Comparison Tool (ACT) (Carver et al. 2005). The sequences have been aligned from the predicted replication origins (oriC; right), with the terminus of replication in the centre. The coloured bars separating each genome (red and blue) represent orthologous matches identified by reciprocal FASTA analysis (Pearson and Lipman 1988). Red lines link orthologues in the same orientation, blue lines link orthologues in the reverse orientation. Variable regions of the chromosomes containing MGEs such as prophages, plasmids, transposons, SaPI and other genomic islands are marked as coloured boxes (see figure for key)

Orthologue comparisons of the chromosomes of S. aureus and related bacteria. Orthologues were identified by reciprocal FASTA analysis (Pearson and Lipman 1988), with an identity cut-off of 30% and a length of match cut-off of 80%. The sequences used for the comparisons were as follows: S. aureus strains MRSA252 (accession number BX571856; Holden et al. 2004) and Mu50 (accession number BA000017; Kuroda et al. 2001), Staphylococcus epidermidis strain RP62a (accession number CP000029; Gill et al. 2005) and Bacillus subtilis strain (accession number AL009126; Kunst et al. 1997). a Pairwise orthologue comparisons of the B. subtilis, S. aureus Mu50, S. aureus MRSA252 and S. epidermidis RP62a chromosomes displayed using the ACT (Carver et al. 2005). The sequences have been aligned from the predicted replication origins (oriC; right), with the terminus of replication in the centre. The coloured bars separating each genome (red and blue) represent orthologous matches identified by reciprocal FASTA analysis (Pearson and Lipman 1988). Red lines link orthologues in the same orientation, blue lines link orthologues in the reverse orientation. b Distribution of orthologues in S. aureus, S. epidermidis RP62a and B. subtilis. Orthologues were identified as described above
To understand the genetic basis for the marked differences in the biology and pathology of S. aureus strains, we must look in more details at the genomic inventories of the sequenced strains. From Fig. 1, it is apparent that the S. aureus genome is clearly made up of two major components: the conserved core genome, which is present in all isolates, and the accessory genome, which is unique or is only found in some of the strains.
S. aureus core genome
Approximately 75% of a S. aureus genome comprises a core component consisting of genes present in all of the strains. Apart from those genes that are essential for growth and survival, there are genes with common species-associated functions, including virulence genes not carried by other staphylococcal species, such as cell surface binding proteins, toxins, exoenzymes and the capsule biosynthetic genes (Lindsay and Holden 2004). Whilst this component would appear to be a stable constituent of the S. aureus genome, the contribution of the genes contained within it to S. aureus strain differences should not be overlooked. Small-scale sequence variation can have marked effects on gene expression and protein function. Genetic diversity in the core genome can therefore account for some of the important phenotypic differences between strains.
Single nucleotide polymorphisms
The DNA encoding much of the S. aureus core genome is highly similar. The average level of nucleotide identity between all orthologous gene pairs was 99.8% when MW2 and MSSA476 were compared, 98.65% for MSSA476 and N315 and 97.7% for MRSA252 and N315 (Ed Feil, personal communication). Much of the sequence divergence in the core genome is due to single nucleotide polymorphisms (SNPs). The functional effect of a polymorphism depends on its nature and position. Many of the SNPs within coding regions will be phenotypically silent, as the nucleotide change introduced will not result in a change in the amino acid encoded (synonymous substitution). Non-synonymous substitutions (substitutions that result in a change in the encoded amino acid) may however contribute to differences in the expression and function of genes, thus creating a mutant allele. SNPs are more likely to have functional effects if they occur in the first or second base pair of a codon. Redundancy in the amino acid coding system means that the nucleotide in the third base position of a codon can often be changed without affecting the amino acid it encodes. The functional effect of the allele may be positive or negative depending on the selective conditions.
The routine use of antibiotics to treat S. aureus infections has exerted a sustained and strong selective pressure on clinical strains. Spontaneous antibiotic resistance mutants have appeared for many commonly used therapeutic agents. Fusidic acid is a potent antibiotic that blocks bacterial protein synthesis by inhibiting the function of elongation factor G (EF-G). Point mutations within the gene encoding EF-G (fusA), result in amino acid changes (non-synonymous substitutions) that confer fusidic acid resistance. Analysis of fusidic acid-resistant clinical strains and mutants isolated under fusidic acid selective pressure in vitro has identified 13 amino acid residues within EF-G, which are targets for amino acid exchanges to generate fusidic acid resistance in S. aureus (Besier et al. 2003). Interestingly, the evolution of fusidic acid resistance is not as straightforward as acquiring a single point mutation in the target protein. Recent studies have shown that point mutations in EF-G also have a fitness cost which may limit their long-term selective advantage (Besier et al. 2005). It has been speculated that secondary compensatory mutations in EF-G may help reduce the detrimental effect of the primary resistance-mediating mutation (Besier et al. 2005).
Using comparative analysis, it is possible to identify mutations that will disrupt the translation of a gene (e.g. nonsense and frameshift mutations), as well as mutations that disrupt the function of the protein (e.g. larger-scale insertions and deletions that alter the number of amino acids in a protein). In S. aureus, the percentage of the genome comprising pseudogenes (MRSA252, 2.5%; MSSA476, 0.8%; COL, 36 1.3%; Mu50, 42 1.5%; N315, 1.1%; MW2, 1.0%) (Gill et al. 2005; Holden et al. 2004; Lerat and Ochman 2005) is similar to the level found in most other sequenced bacterial genomes (Liu et al. 2004). In some bacteria, the accumulation of a large number of pseudogenes is indicative of recent evolutionally change (Cole et al. 2001; Parkhill et al. 2003). In S. aureus, there is no indication from the level of pseudogenes that niche change or host specialization has recently occurred. The pseudogenes identified in the sequenced genomes are more likely to be the product of recent mutational events and therefore may provide clues as to the selective pressures experienced by the strain.
SNPs have also been exploited by multilocus sequence typing (MLST) to investigate the phylogenetic relationships of bacterial strains. MLST is a molecular typing technique exploiting allelic variation in the core genome. The technique is based on the sequence fragments of seven core housekeeping genes (arcC, aroE, glpF, gmk, pta, tpi and yqiL) (Maiden et al. 1998). The stable nature of the core genome and its vertical inheritance mean that allelic variation can be used to measure genetic relatedness. For each S. aureus isolate, these sequences are compared with existing alleles in more than 1,300 S. aureus isolates in the MLST database (http://saureus.mlst.net), generating a numerical allelic profile or sequence type (ST) (Enright et al. 2000). Isolates with STs that have five or more identical alleles out of seven can be assigned to specific clonal complexes (CCs), inferring that these isolates are related and are descendants of a recent clonal ancestor. The method is highly discriminatory, reliable and reproducible, and the data generated have allowed a comprehensive understanding of the diversity of S. aureus populations (Feil et al. 2003). Using MLST, we can compare the sequenced strains to each other and to the wider population and therefore add valuable context to the genomes (see later section for discussion).
Diversity within genes and operons
Genetic diversity in the core genome can extend beyond SNPs, to encompass larger regions of DNA diversity. These can range in size from a few nucleotides within a gene to larger stretches of several kilobase pairs that include complete or partial genes within operons. Sequence comparisons of these regions reveal blocks of divergent sequence, giving the impression of a mosaic genetic structure. The introduction of diversity at this level can have a marked impact on gene function. The apparently mosaic structure of these larger regions, where divergent sequence is flanked by homologous DNA, has led to the suggestion that these regions have arisen by some form of genetic exchange and homologous recombination.
An example of where the replacement of genes within a conserved set of genes can lead to a change in biological properties can be found in the capsule cluster (Sau et al. 1997). S. aureus produces a capsular polysaccharide (CP) that enhances its resistance to host innate immune defences (Thakker et al. 1998). Most clinical isolates of S. aureus are encapsulated, and the serotypes CP5 and CP8 are the most prevalent (O’Riordan and Lee 2004). Significantly, CP5 and CP8 differ in a number of biological properties which are thought to contribute to the relative virulence of serotype CP5 and CP8 S. aureus in vivo (Watts et al. 2005). The gene clusters responsible for the biosynthesis of CP5 and CP8 contain the same number of genes in a single operon and are located at the same site on the chromosome (Sau et al. 1997). However, the two clusters differ in an internal region that contains four capsule type-specific genes (cap5HIJK and cap8HIJK) that exhibit little sequence similarity. Capsules CP5 and CP8 comprise the same trisaccharide repeating units and differ only in the linkages between the sugars and sites of O acetylation (Fournier et al. 1984; Moreau et al. 1990). The capsule-specific genes of the CP5 and CP8 clusters are thought to be responsible for the differing linkages and acetylation of the carbohydrates.
Within the S. aureus genome, there are also examples of apparently chimeric genes, which contain internal regions of variation. The agr cluster contains four genes that encode components of a quorum-sensing system involved in the regulation of virulence (Novick et al. 1995). AgrA and AgrC comprise a two-component signal transduction system (Lina et al. 1998), and AgrB and AgrD are required to produce an autoinducing peptide (AIP) that activates the quorum-sensing system (Ji et al. 1995). AIP is a modified peptide derived from AgrD, which is processed by AgrB (Ji et al. 1997). Four groups of agr systems have been identified on the basis of the ability of their AIP to cross-activate or cross-inhibit agr quorum sensing (Ji et al. 1997; McDowell et al. 2001). The AIP of each group has a different structure. Sequence comparison of the agr loci of these four groups reveals an internal variable region including the C terminus of agrB, the whole of agrD and the N-terminal half of agrC (Dufour et al. 2002b). The covariation of the AIP structural gene and domains of the processing and sensing proteins maintain the functional integrity of each agr system. Replacement of this signal-specific cassette provides a potential means by which strains could swap the specificity of their quorum signals and maintain the induction activity.
Repeat variation
Repeated sequences are widespread in the S. aureus genome. Several studies have identified families of perfect repeats (Baba et al. 2004; Cramton et al. 2000) and tandem repeats (Hardy et al. 2004) predominantly found within intergenic regions. In addition, there are many examples of polymeric tracts and imperfect repeat regions within protein-coding sequences (McDevitt and Foster 1995; Roche et al. 2003). Comparisons of these repeat regions in the different strains show that they can vary in size from a single nucleotide to several hundreds.
An example of how small-scale repeat variation may influence the expression of virulence genes is to be found in the gene encoding a major histocompatibility complex analogue protein (MapW). In six of the seven sequenced strains (N315, MW2, Mu50, COL, NCTC8325 and MSSA476), this gene is truncated due to a frameshift mutation occurring at a poly(A) tract. The repeat in these strains contains either 9 or 10 adenine residues, which introduces a stop codon immediately after it. If the tract contains 8 or 11 residues, the correct frame is maintained and the full-length gene exists. In a random selection of 45 S. aureus strains, the number of adenines in this tract was shown to be varied, with 80% of the strains containing repeats resulting in frameshifts (Buckling et al. 2005). The other 20% of the strains contain either 8 or 11 residues in the tract, thus resulting in a full-length product gene. Protein analysis of strains containing the full-length and truncated forms of the mapW detected both long and short forms of the protein (Buckling et al. 2005). Interestingly, the number of adenines in the repeat can vary in closely related strains, suggesting that phase variation may be occurring. In a clonal bacterial population, phase variation, in which individual cells either express the phase-variable protein or not, results in a heterogenic phenotype (for a review on phase variation, see van der Woude and Baumler 2004). Mechanisms such as slip–strand mispairing (Moxon et al. 1994) may have a role to play in the regulation of this locus.
One group of proteins that contain larger variable repeats is the surface-associated proteins. These proteins play an important role in modulating the interaction between the bacterium and the host and are likely to be immunogenic (Mazmanian et al. 2001; Schneewind et al. 1992). Fibrinogen-binding protein ClfA and collagen-binding protein Cna both contain repetitive protein motifs that have been shown to be important for functional surface expression (Hartford et al. 1997; Hartford et al. 1999). It has been postulated that the repeat regions span the cell wall peptidoglycan and act as a ‘stalk’ to allow the ligand-binding domain of the protein to be displayed in a flexible manner some distance away from the cell surface (Hartford et al. 1997). Many of the other surface-associated proteins also contain repeat regions (e.g. SasA, SasC, SasI) (Roche et al. 2003). The sizes of the repeat regions of some of these proteins have shown to vary between different strains (McDevitt and Foster 1995; Roche et al. 2003). For example, ClfA possesses a dipeptide repeat region composed predominantly of aspartic acid and serine residues. Analysis of this region in several strains showed that the size of the clfA repeat varied from 580 to 1320 bp (McDevitt and Foster 1995). Variation generated by DNA recombination in the regions encoding the repeat regions may represent a means by which S. aureus can evade the host immune system during a long-term infection (Jarraud et al. 2001).
Recombination between repeat regions may also play a role in shaping the inventory of surface proteins in a genome. For example, the locus encoding the bone sialoprotein-binding protein (bbp) and the serine–aspartate repeat proteins sdrD and sdrC contains large regions of repetitive DNA sequence (Roche et al. 2003). In some strains such as MSSA476, all three genes are present, but in MRSA252, sdrD is missing. If the MSSA476 and MRSA252 loci are compared, it is possible to identify homologous DNA sequences in adjacent genes (Fig. 3). For example, the C-terminal region of sdrC is similar to the C-terminal regions of sdrD and bbp. It is therefore possible that the genes in this locus may have originally evolved by gene duplication. In MRSA252, the lack of sdrD is possibly the result of recent gene deletion via homologous recombination between these repeat sequences and the C-terminal regions of sdrD and sdrC (marked in yellow in Fig. 3), resulting in a gene fusion, thus leaving two genes at this locus.

DNA repeat variation in S. aureus surface-associated proteins. Dotplot comparison of the sdr and bbp surface protein loci of MRSA252 and MSSA476. The centre of the figure shows the DNA–DNA similarities between the sdrC and bbp genes of MRSA252 and sdrC, sdrD and bbp genes of MSSA476, with similar sequences represented by dots or lines. The genes are colour-coded to identify the orthologous genes; the N-terminal signal sequence (blue box) and C-terminal transmembrane regions (white box) are marked on the genes. Regions of sequence identity between the orthologous genes are displayed as lines. The serine–aspartate rich repeat regions in the C termini of the surface proteins can be seen as hatched region; the rectangular shape of some of the boxes indicates the variation in the number of repeats between orthologues. The C termini of MSSA476 sdrC and sdrD and MRSA252 sdrC share repeat regions (marked below the genes as yellow boxes). Recombination between the repeats in sdrC and sdrD may be the cause of deletion of sdrD in MRSA252
Accessory genome
The variable, or accessory, component of the S. aureus genome contains genes that encode a diverse array of non-essential functions, ranging from virulence, drug and metal resistance to substrate utilization and miscellaneous metabolism. Many of the regions that make up the accessory genome are mobile genetic elements (MGEs) that can be transferred horizontally between bacteria, including bacteriophage, S. aureus pathogenicity islands (SaPI), staphylococcal cassette chromosomes (SCC), plasmids and transposons (Lindsay and Holden 2004). Interestingly, virulence genes tend to be found on phages and SaPI (Table 1), whilst resistance genes rely on SCC, plasmids and transposons for transfer (Table 2). In the next section of this review, we will focus on the different groups of MGEs found in S. aureus and on the genes carried that are important for transfer and function.
All horizontal gene transfer in bacteria is via three mechanisms: transformation, conjugation and transduction (Thomas and Nielsen 2005). However, S. aureus lack the genes necessary for transformation, and conjugative transfer of elements between S. aureus does not appear to be common. Therefore, most horizontal transfer by S. aureus is probably dependent on transduction by bacteriophage, which therefore plays a pivotal role in the evolution of this pathogen.
Bacteriophage
The horizontal transfer of virulence factors by bacteriophage occurs in two ways: first, by transfer and integration of a bacteriophage that encodes a virulence gene on its genome (phage conversion); second, by generalized transduction.
Many S. aureus bacteriophage genomes contain virulence genes, such as sak (staphylokinase A thrombolytic enzyme; Collen 1998), chp (chemotaxis inhibitory protein and inhibitor of leucocyte migration; de Haas et al. 2004), sea (enterotoxin A, a common cause of food poisoning; Betley and Mekalanos 1985), eta (exfoliative toxin A, a cause of scalded skin syndrome; Yamaguchi et al. 2000) and lukSF-PV (Panton–Valentine leucocidin implicated in haemolytic pneumonia and severe skin and soft tissue infections; Kaneko et al. 1998; Narita et al. 2001). The transfer of S. aureus bacteriophage into and out of isolates probably occurs with significant frequency in nature, including during the course of colonization or infection of patients (Goerke et al. 2004; Moore and Lindsay 2001).
In S. aureus, phages integrated into the chromosome (prophage) are common, with most strains carrying between one and three (Table 1) (Iandolo et al. 2002). These lysogenic phages are “dormant” and are replicated as part of the entire bacterial chromosome and passed to daughter cells during cell division (vertical transfer). Prophages are induced by stress, such as UV light, DNA-damaging agents and some antibiotics, most likely via the bacterial SOS response (Lindsay et al. 1998; Ubeda et al. 2005). During induction, the phage is excised from the chromosome and begins replicating. As phage genomes multiply, the proteins necessary for phage head and tail structures are produced, and the phage genomes are packaged. Eventually, the bacterial cell will lyse, releasing infectious phage particles. These phages then bind to recipient S. aureus cells via an unknown receptor, delivering the phage genome to the cell. The phage may then enter a lytic cycle and lead to the production of more infectious phage or enter a lysogenic cycle and integrate its genome into the bacterial chromosome. An interesting consequence of the induction of phage and phage genome replication is the increased copy number of any phage-carried toxin genes, leading to enhanced toxin production (Sumby and Waldor 2003).
The S. aureus phage genomes contain an integrase gene carried at one end of the genome, and some also have a putative excisionase gene. All have an integrase promotor region similar to the lambda phage model (Carroll et al. 1995), which is therefore predicted to control the lytic/lysogenic switch. It also suggests that a bacterial cell carrying a phage (lysogen) will be immune to infection with a similar phage, but this remains to be proven. S. aureus phage also appear to integrate at only a single site, and this is due to specificity of the integrase and the phage genome’s left and right junctions (Lee and Iandolo 1986a). At least two S. aureus phage integration sites are within known virulence genes, β-haemolysin (hlb) and lipase (geh), and thus, phage conversion leads to loss of these gene products (Coleman et al. 1989; Lee and Iandolo 1986b).
Generalized transduction in S. aureus is thought to be widespread and is the only feasible mechanism for the horizontal transfer of many non-phage MGEs. Only one naturally occurring generalized transducing phage (ϕ11) (Novick 1963) has been well studied, although how it “accidentally” packages up to 45 kb of bacterial DNA instead of its own phage genome is not clear. The resulting phage heads are able to deliver this DNA to other S. aureus cells. Once delivered, the DNA will only survive if it can replicate or integrate into a replicon such as chromosome or a plasmid. For normal chromosomal DNA with no encoded replication mechanism, the only chance of survival is to recombine with host chromosome via homologous recombination. This process is very inefficient in S. aureus. Nevertheless, in the laboratory setting, generalized transduction is the most efficient mechanism for moving DNA between S. aureus isolates.
S. aureus pathogenicity islands
S. aureus pathogenicity islands (SaPI) encode a number of virulence genes, including the superantigens, toxic shock syndrome toxin-1 (tst) and enterotoxins B and C (seb, sec) (Table 1). Superantigens are so called as they can elicit a non-specific T-cell response, leading to massive cell expansion and shock. However, superantigen genes are widespread in S. aureus, and shock is a rare complication of S. aureus colonization or infection. In the sequenced S. aureus strains, six SaPI have been identified, four of which carry superantigen genes (Table 1).
SaPI transfer horizontally in the presence of ‘helper’ phage (Lindsay et al. 1998). SaPI themselves are approximately 15 kb and encode an integrase at one end, similar to the bacteriophage integrases. The integrase promotor region is also similar to that of phage, and SaPI also appear to integrate site specifically (Lindsay et al. 1998). However, they do not encode other genes for the actual transfer process and rely on phage genes for induction. The process can be triggered by infection of the bacterial cell with the helper phage or induction of a helper prophage using stress (Lindsay et al. 1998). The SaPI is excised and packaged into miniature phage-like particles with heads and tails at the same time that normal-sized infectious phage particles are produced (Ruzin et al. 2001). The SaPI particles deliver the SaPI DNA to new S. aureus recipient cells, and the SaPI DNA integrates into the recipient chromosome. This process is extremely efficient because the SaPI-infected cells are not infected with a viable bacteriophage that often causes cell lysis (Lindsay et al. 1998).
Staphylococcal cassette chromosomes
Staphylococcal cassette chromosomes (SCC) always insert into a specific region of the S. aureus chromosome, approximately 25 kb from the origin of replication. SCC often encode antibiotic resistance genes, such as the mec operon for methicillin resistance (SCCmec) (Ito et al. 2001) and far for fusidic acid resistance (SCCfar) (Holden et al. 2004). At least five different versions of SCCmec are found in S. aureus, SCCmec types I to V (Ito et al. 2001, 2004; Ma et al. 2002), three of which are found in the sequenced strains (Table 2). SCC frequently include transposons and integrated plasmids which carry other resistances such as kanamycin and erythromycin (Hiramatsu et al. 2001). Genes responsible for a particularly mucoid version of the S. aureus capsule have also been reported on SCC (Luong et al. 2002).
SCC vary substantially in length and look like a mosaic structure of integrations and recombinations with various genetic fragments (Ito et al. 2001; Ito et al. 2004; Ma et al. 2002). Typically, SCC carry one (ccrC) (Ito et al. 2004) or two ccr genes (ccrA and ccrB) (Katayama et al. 2000) that encode site-specific recombinases, which catalyse the excision and integration of a circular version of the SCC element. How the element replicates and then transfers horizontally between bacteria is not clear. Since the original SCC are too large to fit into a generalized transducing phage head, this may account for why SCCmec spread at relatively low frequency. However, the new SCCmec type IVs appear to spread much more easily between isolates (Ma et al. 2002; Robinson and Enright 2003); this could be because of their smaller size, which allows them to be transduced.
Plasmids
There are three families of S. aureus plasmid based on size and ability to conjugate (Paulsen et al. 1997). Class I are the smallest and have the highest copy number, whilst only class III are conjugative. Class I are usually less than 5 kb, found at high copy number and encode only one or two resistance genes (Table 2), such as pT181 encoding tetracycline resistance in COL (Gill et al. 2005; Khan and Novick 1983). In some cases, class I plasmids can also be integrated into the chromosome; pUB110 carrying kanamycin and bleomycin resistance is integrated into the type II SCCmec elements found in three of the sequenced strains (Holden et al. 2004; Ito et al. 2001; Kuroda et al. 2001) (Table 2). Class I plasmids can be transferred between S. aureus by generalized transduction at approximately 100 times the frequency of chromosomal markers (Ubelaker and Rosenblum 1978). This could be due to plasmid concatemers that preferentially package into phage heads (Dyer et al. 1985). They can also be mobilized by some class III conjugative plasmids at low frequency (Projan and Archer 1989).
Class II plasmids are up to 40 kb and typically carry resistance to one or more of the β-lactams (bla), heavy metals (ars, cad, mer), antiseptics (qac) or aminoglycosides (aacA–aphD) (Table 2). The resistance genes are often on transposons that have integrated into the plasmid, and plasmid variants are often due to rearrangements of these elements. It is likely that class II plasmids are transferred between S. aureus by generalized transduction.
Class III plasmids are similar to the class II plasmids, but also encode transfer (tra) genes that allow conjugative transfer of the plasmid between bacterial isolates at low frequency on solid surfaces (Thomas and Archer 1989) (Table 2). Since some class III plasmids are up to 60 kb, they can be too large to be transferred by transduction.
Transposons
S. aureus transposons often encode resistance genes. For example, Tn554 encodes resistance to erythromycin (Phillips and Novick 1979) and is often associated with type II SCCmec. Once a strain is positive for this transposon, multiple copies can become integrated at various sites around the chromosome (Table 2). Tn552 encodes β-lactamase resistance and is often associated with class II and III plasmids, but can also be found in the chromosome (Rowland and Dyke 1989). Tn5801 is a large transposon integrated into the Mu50 genome (Table 2) and encodes tetracycline resistance (tetM) (Kuroda et al. 2001). Many other transposons have also been described.
All the transposons encode a transposase gene, and the product of this gene catalyses excision and/or replication of the element, as well as integration. Horizontal transfer of transposons to other S. aureus cells is presumably by “piggybacking” onto another MGE that is transferred, most likely a plasmid. The plasmid can then be transferred by transduction or conjugation. We are unaware of any reports of a transposon integrating into a phage or SaPI.
Conjugative transposons have been described in S. aureus; they are transposons that include tra genes, but they cannot replicate autonomously. Examples are Tn916 and Tn918, originally from streptococci (Clewell et al. 1985). However, it is not clear if native conjugative transposons are found in S. aureus. Interestingly, the genomes of MRSA252 and COL contain novel regions containing genes that have weak similarity to Tn916, which could be a related element (Table 2).
The first fully vancomycin-resistant strains of S. aureus have now been described (Anonymous 2002a,b, 2004), and if these isolates were to spread in hospitals, they would cause a serious treatment dilemma. The gene for resistance, vanA, is encoded on a transposon derived from enterococci (Weigel et al. 2003). The likely mechanism of transfer was that the transposon in the enterococcus jumped onto a pheromone-responsive conjugative plasmid, which then transferred via conjugation to S. aureus. The plasmid was unable to replicate in S. aureus, but the transposon had the opportunity to jump into the S. aureus chromosome and/or a resident S. aureus plasmid (Clewell et al. 1985; Weigel et al. 2003).
Genomic islands and islets
In addition to the previously characterized MGEs, there are regions of the S. aureus chromosome that have been designated as genomic islands, which are thought to have arisen by horizontal gene transfer. The origins and modes of transfer remain unclear; however, the compositions of these regions vary between strains and often contain genes associated with pathogenicity. Two such regions (Kuroda et al. 2001) are νSaα, encoding multiple exotoxins (set) (Jarraud et al. 2001; Williams et al. 2000) and lipoproteins (lpl), and νSaβ, encoding multiple set and serine protease homologues (spl) (Reed et al. 2001). Both these genomic islands seem extremely stable as they are found in all sequenced isolates (Table 1) in the same location but exhibit allelic variation (Lindsay and Holden 2004). For example, in νSaα, the number of set and lpl varies between strains: MRSA252 contains nine set and seven putative lpl, one of which is a pseudogene; MSS476 contains eleven set and five putative lpl. νSaβ is more diverse than νSaα: MRSA252 contains five spl, two of which are pseudogenes, and six set; MSSA476 contains four spl, two leucotoxin homologues (lukDE) (Gravet et al. 1998) and a lantibitic biosynthesis cluster (bsa). In addition, the MRSA252 island also contains a hyaluronate lysase paralogue (hysA), with 75% identity at the amino acid level to a hyaluronate lysase found elsewhere on the chromosome (SAR2292) (Farrell et al. 1995).
Horizontal gene transfer in other bacteria
The diversity of the MGEs in S. aureus suggests that it has catholic taste, utilizing different classes of elements to augment the virulence and drug resistance components of its genome. This raises the question as to what drives the species to be so varied and accommodating in its evolution. For some bacterial pathogens, strain diversity can be reliant on a particular type of MGE or mechanism of genetic exchange. For example, Streptococcus pyogenes (alternatively referred to as group A Streptococcus) is responsible for a diverse number of diseases in humans, including pharyngitis, acute rheumatic fever (ARF), toxic shock syndrome (TSS), impetigo and scarlet fever. The genomes of five different S. pyogenes strains have been sequenced, and the main source of diversity between the genomes is mainly due to the presence of different prophages. Each strain contains between four and six prophages or prophage-like elements, and each of these MGEs contains characterized or putative virulence factors (Banks et al. 2004; Beres et al. 2004; Ferretti et al. 2001; Nakagawa et al. 2003; Smoot et al. 2002). Like S. aureus, S. pyogenes has a close association with the human host and also has the potential to evolve new variants with increased virulence and drug resistance. Why, therefore, has S. pyogenes taken the route of poly-lysogeny to be the main source of strain variation, whereas S. aureus is more eclectic? The answer to this question perhaps lies in acquiring a greater understanding of the diversity of the accessory genome of a species.
Are six S. aureus genomes enough?
Whilst the increasing multiplicity of genome sequences provides an insight into the sets of genes carried by individual species and the diversity of MGEs, it can also mislead. The apparent genetic make-up of a species can also be distorted by the choice of strains sequenced. Often, they are strains chosen that are deemed to be important for some biological or political reason, for example, epidemic strains, laboratory strains and strains associated with a distinct pathology. However, the strains chosen may not be typical of the species as a whole. This is illustrated by the S. aureus strains thus far sequenced. S. aureus is a commensal bacterium that is carried by a large proportion of the population. For the vast majority of individuals, carriage will not cause any problems and they will be asymptomatic. The population of S. aureus is not only confined to humans; S. aureus are carried by, and can cause disease in, animals (van Duijkeren et al. 2004; Vautor et al. 2005). However, our knowledge of the S. aureus genome is shaped by the sequence of five human disease-causing isolates (N315, Mu50, MW2, MRSA252 and MSSA476), an MRSA relic (COL) and a laboratory strain (NCTC8325). This therefore raises the question of how representative the genomes are in comparison to carriage strains and the wider population of S. aureus.
Using MLST, it is possible to investigate evolutionary relationships of the genome sequence strains in relation to each other and representatives of the wider population. Figure 4 shows a population snapshot of STs in the MLST database generated using eBURST (Feil et al. 2004). S. aureus has a clonal population structure (Enright et al. 2002; Feil et al. 2003). In a study of 334 isolates, including carriage and clinical strains, 87% of S. aureus in hospitals and the community belong to any one of 11 CCs (Fig. 4) (Feil et al. 2003). The seven sequenced strains belong to four of these CCs: both the hospital-acquired strains N315 and Mu50 and community-acquired strains MW2 and MSSA476 belong to identical but separate STs (ST5 and ST1) in different CCs. COL and NCTC8325 belong to closely related STs (ST250 and ST8) but in the same CC. MRSA252 belongs to CC30 and appears to be the most divergent of the sequenced strains (ST36). Comparative genomic analysis revealed that about 6% of the genome is novel in comparison to other sequenced strains (Holden et al. 2004). Using the population framework as a guide, it is possible to see that the apparent diversity of MRSA252 is due to the relatedness of the other sequenced genomes rather than any true divergence. If a fuller picture of the S. aureus genome is to be gained, strains belonging to the other CCs and non-CCs should be sequenced.

Population snapshot of S. aureus. Clusters of related sequence types (STs) and individual unlinked STs within the entire S. aureus multilocus sequence typing (MLST) database (http://saureus.mlst.net/) are displayed as a single eBURST (Feil et al. 2004) diagram by setting the group definition to zero of seven shared alleles. The figure was generated from 1,360 isolates, corresponding to 372 STs, in the S. aureus MLST database. Clusters of linked isolates correspond to CCs. Primary founders (blue) are positioned centrally in the cluster, and subgroup founders are shown in yellow. The eight major and three minor CCs described by Feil et al. (2003) are indicated; the ST labels have been removed for clarity. The position of the seven sequenced strains are indicated in red, with their ST indicated in brackets
Future evolution of pathogenic and drug-resistant strains
Many MGEs encode genes that contribute to S. aureus virulence and pathogenesis, and they can be transferred between S. aureus isolates. This transfer leads to the evolution of S. aureus causing novel clinical challenges. For example, SCCmec encoding methicillin resistance are now found in S. aureus that are particularly adapted to surviving and spreading in hospitals (Moore and Lindsay 2002). Other examples are the community-acquired MRSAs (positive for SCCmec) that also carry lukSF-PV on a bacteriophage, which seem to spread and cause skin and soft tissue infection in otherwise healthy people (Dufour et al. 2002a). A further example is the recent emergence of three isolates of vancomycin-resistant MRSA, which are positive for SCCmec and the vanA transposon (Weigel et al. 2003).
It therefore seems likely that these MGEs will be capable of transferring further, potentially generating strains of S. aureus that are resistant to methicillin and vancomycin and capable of spreading in hospitals and causing disease in healthy patients.
Despite the widespread distribution of S. aureus and serious antibiotic selective pressure, such strains have not been identified as yet. A likely explanation is that there are barriers to horizontal transfer and that these are efficient in nature. For example, S. aureus have restriction–modification (RM) pathways (Iordanescu and Surdeanu 1976), which probably inhibit horizontal transfer. Restriction enzymes digest DNA at specific sequences, and modification enzymes modify those sequences in the bacterium’s own DNA, preventing restriction. Thus, RM should prevent the horizontal transfer of MGE from “foreign” bacteria. The genome sequencing projects identify at least two S. aureus-encoded type I RM genes on the chromosome and genomic islands (Holden et al. 2004; Kuroda et al. 2001), and gene variation between sequenced strains occurs. More recently, Dempsey et al. (2005) have identified RM genes in a S. aureus bacteriophage, which is capable of conferring resistance to lysis by other bacteriophage.
Other barriers to horizontal transfer may also be important. It is thought that an isolate lysogenic for a bacteriophage is resistant to infection with a related bacteriophage (bacteriophage immunity), and a similar mechanism may be functioning in SaPI. Plasmids can be classed into different “incompatibility” groups, such that two plasmids with the same replication mechanism cannot survive in the same cell (Novick 1987). A further explanation is that much horizontal transfer is dependent on generalized transduction by bacteriophage, but the distribution and frequency of such phages are not known. Further investigation of horizontal transfer of MGE is essential to truly understand S. aureus evolution and the potential for more virulent and resistant strains to emerge.
In addition to the barriers associated with the mode of transport of MGEs, there is growing evidence that the genetic background of a recipient can also contribute to the success of a transfer. The distribution of methicillin resistance conferring SCCmec elements is not uniform in the population; not all the major CCs of S. aureus contain a high frequency of MRSA strains (Enright et al. 2002). It has been hypothesized that some genetic backgrounds may be restrictive of the methicillin resistance gene (mecA) and its expression, which could account for the restricted clonal distribution of SCCmec in nature (Katayama et al. 2005). When mecA was introduced on a low-copy-number plasmid into methicillin-susceptible S. aureus clinical isolates, the transformability and stability of the mecA plasmid were greater in strains from the CCs that contain higher numbers of MRSA strains (Katayama et al. 2005). As far as SCCmec is concerned, it would therefore appear that all S. aureus are not equal; some innate genetic or biochemical property of the strains influences the success of SCCmec.
In summary, S. aureus is a bacterium that typically lives in the human nose and has evolved with us. Approximately 11 clonal lineages dominate, and it remains to be seen what gene combinations and variations make these lineages so successful. In addition, MGEs move in and out of S. aureus isolates with significant frequency. The accumulation of MGEs in some isolates makes them increasingly virulent and resistant to antibiotics and able to cause new clinical challenges. Our current understanding suggests that we may have seen only “the tip of the iceberg” and that S. aureus will continue to evolve, potentially generating isolates that can create havoc in our hospitals as well as in healthy patients outside of the healthcare setting.
References
Anonymous (2002a) Staphylococcus aureus resistant to vancomycin—United States, 2002. MMWR Morb Mortal Wkly Rep 51:565–567
Anonymous (2002b) Vancomycin-resistant Staphylococcus aureus—Pennsylvania, 2002. MMWR Morb Mortal Wkly Rep 51:902
Anonymous (2004) Vancomycin resistant Staphylococcus aureus—New York, 2004. MMWR Morb Mortal Wkly Rep 53:322–323
Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, Nagai Y, Iwama N, Asano K, Naimi T, Kuroda H, Cui L, Yamamoto K, Hiramatsu K (2002) Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359:1819–1827
Baba T, Takeuchi F, Kuroda M, Ito T, Yuzawa H, Hiramatsu K (2004) The Staphylococcus aureus geneome. In: Ala’Aldeen D, Hiramatsu K (eds) Staphylococcus aureus molecular and clinical aspects. Horwood Publishing, Chichester, UK, pp 66–153
Banks DJ, Porcella SF, Barbian KD, Beres SB, Philips LE, Voyich JM, DeLeo FR, Martin JM, Somerville GA, Musser JM (2004) Progress toward characterization of the group A Streptococcus metagenome: complete genome sequence of a macrolide-resistant serotype M6 strain. J Infect Dis 190:727–738
Battistuzzi FU, Feijao A, Hedges SB (2004) A genomic timescale of prokaryote evolution: insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol Biol 4:44
Beres SB, Sylva GL, Sturdevant DE, Granville CN, Liu MY, Ricklefs SM, Whitney AR, Parkins LD, Hoe NP, Adams GJ, Low DE, DeLeo FR, McGeer A, Musser JM (2004) Genome-wide molecular dissection of serotype M3 group A Streptococcus strains causing two epidemics of invasive infections. Proc Natl Acad Sci U S A 101:11833–11838
Besier S, Ludwig A, Brade V, Wichelhaus TA (2003) Molecular analysis of fusidic acid resistance in Staphylococcus aureus. Mol Microbiol 47:463–469
Besier S, Ludwig A, Brade V, Wichelhaus TA (2005) Compensatory adaptation to the loss of biological fitness associated with acquisition of fusidic acid resistance in Staphylococcus aureus. Antimicrob Agents Chemother 49:1426–1431
Betley MJ, Mekalanos JJ (1985) Staphylococcal enterotoxin A is encoded by phage. Science 229:185–187
Bolotin A, Quinquis B, Renault P, Sorokin A, Ehrlich SD, Kulakauskas S, Lapidus A, Goltsman E, Mazur M, Pusch GD, Fonstein M, Overbeek R, Kyprides N, Purnelle B, Prozzi D, Ngui K, Masuy D, Hancy F, Burteau S, Boutry M, Delcour J, Goffeau A, Hols P (2004) Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus. Nat Biotechnol 22:1554–1558
Buckling A, Neilson J, Lindsay J, ffrench-Constant R, Enright M, Day N, Massey RC (2005) Clonal distribution and phase-variable expression of a major histocompatibility complex analogue protein in Staphylococcus aureus. J Bacteriol 187:2917–2919
Carroll D, Kehoe MA, Cavanagh D, Coleman DC (1995) Novel organization of the site-specific integration and excision recombination functions of the Staphylococcus aureus serotype-F virulence-converting phages phi-13 and phi-42. Mol Microbiol 16:877–893
Carver TJ, Rutherford K, Berriman M, Rajandream MA, Barrell B, Parkhill J (2005) ACT: the Artemis Comparison Tool. Bioinformatics 21:3422–3423
Cazalet C, Rusniok C, Bruggemann H, Zidane N, Magnier A, Ma L, Tichit M, Jarraud S, Bouchier C, Vandenesch F, Kunst F, Etienne J, Glaser P, Buchrieser C (2004) Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat Genet 36:1165–1173
Chambers HF (2005) Community-associated MRSA—resistance and virulence converge. N Engl J Med 352:1485–1487
Chang S, Sievert DM, Hageman JC, Boulton ML, Tenover FC, Downes FP, Shah S, Rudrik JT, Pupp GR, Brown WJ, Cardo D, Fridkin SK (2003) Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N Engl J Med 348:1342–1347
Clewell DB, An FY, White BA, Gawronburke C (1985) Streptococcus faecalis sex-pheromone (cAM373) also produced by Staphylococcus aureus and identification of a conjugative transposon (Tn918). J Bacteriol 162:1212–1220
Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honore N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holroyd S, Hornsby T, Jagels K, Lacroix C, Maclean J, Moule S, Murphy L, Oliver K, Quail MA, Rajandream MA, Rutherford KM, Rutter S, Seeger K, Simon S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Taylor K, Whitehead S, Woodward JR, Barrell BG (2001) Massive gene decay in the leprosy bacillus. Nature 409:1007–1011
Coleman DC, Sullivan DJ, Russell RJ, Arbuthnott JP, Carey BF, Pomeroy HM (1989) Staphylococcus aureus bacteriophages mediating the simultaneous lysogenic conversion of beta-lysin, staphylokinase and enterotoxin-A—molecular mechanism of triple conversion. J Gen Microbiol 135:1679–1697
Collen D (1998) Staphylokinase: a potent, uniquely fibrin-selective thrombolytic agent. Nat Med 4:279–284
Cosgrove SE, Sakoulas G, Perencevich EN, Schwaber MJ, Karchmer AW, Carmeli Y (2003) Comparison of mortality associated with methicillin-resistant and methicillin-susceptible Staphylococcus aureus bacteremia: a meta-analysis. Clin Infect Dis 36:53–59
Cramton SE, Schnell NF, Gotz F, Bruckner R (2000) Identification of a new repetitive element in Staphylococcus aureus. Infect Immun 68:2344–2348
de Haas CJC, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJB, Heezius E, Poppelier M, Van Kessel KPM, van Strijp JAG (2004) Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med 199:687–695
Dempsey RM, Carroll D, Kong H, Higgins L, Keane CT, Coleman DC (2005) Sau42I, a BcgI-like restriction–modification system encoded by the Staphylococcus aureus quadruple-converting phage Phi42. Microbiology 151:1301–1311
Dufour P, Gillet Y, Bes M, Lina G, Vandenesch F, Floret D, Etienne J, Richet H (2002a) Community-acquired methicillin-resistant Staphylococcus aureus infections in France: emergence of a single clone that produces Panton–Valentine leukocidin. Clin Infect Dis 35:819–824
Dufour P, Jarraud S, Vandenesch F, Greenland T, Novick RP, Bes M, Etienne J, Lina G (2002b) High genetic variability of the agr locus in Staphylococcus species. J Bacteriol 184:1180–1186
Dyer DW, Rock MI, Lee CY, Iandolo JJ (1985) Generation of transducing particles in Staphylococcus aureus. J Bacteriol 161:91–95
Emmerson AM, Enstone JE, Griffin M, Kelsey MC, Smyth ET (1996) The Second National Prevalence Survey of infection in hospitals—overview of the results. J Hosp Infect 32:175–190
Enright MC, Day NPJ, Davies CE, Peacock SJ, Spratt BG (2000) Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol 38:1008–1015
Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG (2002) The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc Natl Acad Sci U S A 99:7687–7692
Farrell AM, Taylor D, Holland KT (1995) Cloning, nucleotide sequence determination and expression of the Staphylococcus aureus hyaluronate lysase gene. FEMS Microbiol Lett 130:81–85
Feil EJ, Cooper JE, Grundmann H, Robinson DA, Enright MC, Berendt T, Peacock SJ, Smith JM, Murphy M, Spratt BG, Moore CE, Day NPJ (2003) How clonal is Staphylococcus aureus? J Bacteriol 185:3307–3316
Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG (2004) eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol 186:1518–1530
Ferretti JJ, McShan WM, Ajdic D, Savic DJ, Savic G, Lyon K, Primeaux C, Sezate S, Suvorov AN, Kenton S, Lai HS, Lin SP, Qian YD, Jia HG, Najar FZ, Ren Q, Zhu H, Song L, White J, Yuan XL, Clifton SW, Roe BA, McLaughlin R (2001) Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc Natl Acad Sci U S A 98:4658–4663
Fluit AC, Verhoef J, Schmitz FJ (2001) Frequency of isolation and antimicrobial resistance of Gram-negative and Gram-positive bacteria from patients in intensive care units of 25 European university hospitals participating in the European arm of the SENTRY Antimicrobial Surveillance Program 1997–1998. Eur J Clin Microbiol Infect Dis 20:617–625
Fournier JM, Vann WF, Karakawa WW (1984) Purification and characterization of Staphylococcus aureus Type-8 capsular polysaccharide. Infect Immun 45:87–93
Fouts DE, Mongodin EF, Mandrell RE, Miller WG, Rasko DA, Ravel J, Brinkac LM, DeBoy RT, Parker CT, Daugherty SC, Dodson RJ, Durkin AS, Madupu R, Sullivan SA, Shetty JU, Ayodeji MA, Shvartsbeyn A, Schatz MC, Badger JH, Fraser CM, Nelson KE (2005) Major structural differences and novel potential virulence mechanisms from the genomes of multiple Campylobacter species. PLoS Biol 3:72–85
Gill SR, Fouts DE, Archer GL, Mongodin EF, DeBoy RT, Ravel J, Paulsen IT, Kolonay JF, Brinkac L, Beanan M, Dodson RJ, Daugherty SC, Madupu R, Angiuoli SV, Durkin AS, Haft DH, Vamathevan J, Khouri H, Utterback T, Lee C, Dimitrov G, Jiang LX, Qin HY, Weidman J, Tran K, Kang K, Hance IR, Nelson KE, Fraser CM (2005) Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol 187:2426–2438
Gillet Y, Issartel B, Vanhems P, Fournet JC, Lina G, Bes M, Vandenesch F, Piemont Y, Brousse N, Floret D, Etienne J (2002) Association between Staphylococcus aureus strains carrying gene for Panton–Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet 359:753–759
Goerke C, Papenberg SMY, Dasbach S, Dietz K, Ziebach R, Kahl BC, Wolz C (2004) Increased frequency of genomic alterations in Staphylococcus aureus during chronic infection is in part due to phage mobilization. J Infect Dis 189:724–734
Gravet A, Colin DA, Keller D, Girardot R, Monteil H, Prevost G (1998) Characterization of a novel structural member, LukE–LukD, of the bicomponent staphylococcal leucotoxins family. FEBS Lett 436:202–208
Hacker J, Carniel E (2001) Ecological fitness, genomic islands and bacterial pathogenicity—a Darwinian view of the evolution of microbes. EMBO Rep 2:376–381
Hardy KJ, Ussery DW, Oppenheim BA, Hawkey PM (2004) Distribution and characterization of staphylococcal interspersed repeat units (SIRUs) and potential use for strain differentiation. Microbiology 150:4045–4052
Hartford O, Francois P, Vaudaux P, Foster TJ (1997) The dipeptide repeat region of the fibrinogen-binding protein (clumping factor) is required for functional expression of the fibrinogen-binding domain on the Staphylococcus aureus cell surface. Mol Microbiol 25:1065–1076
Hartford O, McDevitt D, Foster TJ (1999) Matrix-binding proteins of Staphylococcus aureus: functional analysis of mutant and hybrid molecules. Microbiology 145:2497–2505
Herold BC, Immergluck LC, Maranan MC, Lauderdale DS, Gaskin RE, Boyle-Vavra S, Leitch CD, Daum RS (1998) Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA 279:593–598
Hiramatsu K, Cui L, Kuroda M, Ito T (2001) The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol 9:486–493
Holden MTG, Feil EJ, Lindsay JA, Peacock SJ, Day NPJ, Enright MC, Foster TJ, Moore CE, Hurst L, Atkin R, Barron A, Bason N, Bentley SD, Chillingworth C, Chillingworth T, Churcher C, Clark L, Corton C, Cronin A, Doggett J, Dowd L, Feltwell T, Hance Z, Harris B, Hauser H, Holroyd S, Jagels K, James KD, Lennard N, Line A, Mayes R, Moule S, Mungall K, Ormond D, Quail MA, Rabbinowitsch E, Rutherford K, Sanders M, Sharp S, Simmonds M, Stevens K, Whitehead S, Barrell BG, Spratt BG, Parkhill J (2004) Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci U S A 101:9786–9791
Iandolo JJ, Worrell V, Groicher KH, Qian YD, Tian RY, Kenton S, Dorman A, Ji HG, Lin SP, Loh P, Qi SL, Zhu H, Roe BA (2002) Comparative analysis of the genomes of the temperate bacteriophages phi 11, phi 12 and phi 13 of Staphylococcus aureus 8325. Gene 289:109–118
Iordanescu S, Surdeanu M (1976) Two restriction and modification systems in Staphylococcus aureus NCTC8325. J Gen Microbiol 96:277–281
Ito T, Katayama Y, Asada K, Mori N, Tsutsumimoto K, Tiensasitorn C, Hiramatsu K (2001) Structural comparison of three types of staphylococcal cassette chromosome mec integrated in the chromosome in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 45:1323–1336
Ito T, Ma XX, Takeuchi F, Okuma K, Yuzawa H, Hiramatsu K (2004) Novel type V staphylococcal cassette chromosome mec driven by a novel cassette chromosome recombinase, ccrC. Antimicrob Agents Chemother 48:2637–2651
Jarraud S, Peyrat MA, Lim A, Tristan A, Bes M, Mougel C, Etienne J, Vandenesch F, Bonneville M, Lina G (2001) egc, a highly prevalent operon of enterotoxin gene, forms a putative nursery of superantigens in Staphylococcus aureus. J Immunol 166:669–677
Ji GY, Beavis R, Novick RP (1997) Bacterial interference caused by autoinducing peptide variants. Science 276:2027–2030
Ji GY, Beavis RC, Novick RP (1995) Cell density control of staphylococcal virulence mediated by an octapeptide pheromone. Proc Natl Acad Sci U S A 92:12055–12059
Johnson AP, Aucken HM, Cavendish S, Ganner M, Wale MCJ, Warner M, Livermore DM, Cookson BD (2001) Dominance of EMRSA-15 and -16 among MRSA causing nosocomial bacteraemia in the UK: analysis of isolates from the European Antimicrobial Resistance Surveillance System (EARSS). J Antimicrob Chemother 48:143–144
Jones RN (2003) Global epidemiology of antimicrobial resistance among community-acquired and nosocomial pathogens: a five-year summary from the SENTRY Antimicrobial Surveillance Program (1997–2001). Semin Respir Crit Care Med 24:121–134
Kaneko J, Kimura T, Narita S, Tomita T, Kamio Y (1998) Complete nucleotide sequence and molecular characterization of the temperate staphylococcal bacteriophage phi PVL carrying Panton–Valentine leukocidin genes. Gene 215:57–67
Katayama Y, Ito T, Hiramatsu K (2000) A new class of genetic element, staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 44:1549–1555
Katayama Y, Robinson DA, Enright MC, Chambers HF (2005) Genetic background affects stability of mecA in Staphylococcus aureus. J Clin Microbiol 43:2380–2383
Khan SA, Novick RP (1983) Complete nucleotide-sequence of pT181, a tetracycline-resistance plasmid from Staphylococcus aureus. Plasmid 10:251–259
Kim T, Oh PI, Simor AE (2001) The economic impact of methicillin-resistant Staphylococcus aureus in Canadian hospitals. Infect Control Hosp Epidemiol 22:99–104
Kluytmans J, vanBelkum A, Verbrugh H (1997) Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev 10:505–520
Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi SK, Codani JJ, Connerton IF, Cummings NJ, Daniel RA, Denizot F, Devine KM, Dusterhoft A, Ehrlich SD, Emmerson PT, Entian KD, Errington J, Fabret C, Ferrari E, Foulger D, Fritz C, Fujita M, Fujita Y, Fuma S, Galizzi A, Galleron N, Ghim SY, Glaser P, Goffeau A, Golightly EJ, Grandi G, Guiseppi G, Guy BJ, Haga K, Haiech J, Harwood CR, Henaut A, Hilbert H, Holsappel S, Hosono S, Hullo MF, Itaya M, Jones L, Joris B, Karamata D, Kasahara Y, KlaerrBlanchard M, Klein C, Kobayashi Y, Koetter P, Koningstein G, Krogh S, Kumano M, Kurita K, Lapidus A, Lardinois S, Lauber J, Lazarevic V, Lee SM, Levine A, Liu H, Masuda S, Mauel C, Medigue C, Medina N, Mellado RP, Mizuno M, Moestl D, Nakai S, Noback M, Noone D, Oreilly M, Ogawa K, Ogiwara A, Oudega B, Park SH, Parro V, Pohl TM, Portetelle D, Porwollik S, Prescott AM, Presecan E, Pujic P, Purnelle B et al (1997) The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390:249–256
Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui LZ, Oguchi A, Aoki K, Nagai Y, Lian JQ, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K (2001) Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 357:1225–1240
Lee CY, Iandolo JJ (1986a) Integration of Staphylococcal phage L54a occurs by site-specific recombination—structural-analysis of the attachment sites. Proc Natl Acad Sci U S A 83:5474–5478
Lee CY, Iandolo JJ (1986b) Lysogenic conversion of staphylococcal lipase is caused by insertion of the bacteriophage L54a genome into the lipase structural gene. J Bacteriol 166:385–391
Lerat E, Ochman H (2005) Recognizing the pseudogenes in bacterial genomes. Nucleic Acids Res 33:3125–3132
Lina G, Jarraud S, Ji GY, Greenland T, Pedraza A, Etienne J, Novick RP, Vandenesch F (1998) Transmembrane topology and histidine protein kinase activity of AgrC, the agr signal receptor in Staphylococcus aureus. Mol Microbiol 28:655–662
Lindsay JA, Holden MTG (2004) Staphylococcus aureus: superbug, super genome? Trends Microbiol 12:378–385
Lindsay JA, Ruzin A, Ross HF, Kurepina N, Novick RP (1998) The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol Microbiol 29:527–543
Liu Y, Harrison PM, Kunin V, Gerstein M (2004) Comprehensive analysis of pseudogenes in prokaryotes: widespread gene decay and failure of putative horizontally transferred genes. Genome Biol 5:R64
Livermore DM (2003) Bacterial resistance: origins, epidemiology, and impact. Clin Infect Dis 36:S11–S23
Luong TT, Ouyang S, Bush K, Lee CY (2002) Type 1 capsule genes of Staphylococcus aureus are carried in a staphylococcal cassette chromosome genetic element. J Bacteriol 184:3623–3629
Ma XX, Ito T, Tiensasitorn C, Jamklang M, Chongtrakool P, Boyle-Vavra S, Daum RS, Hiramatsu K (2002) Novel type of staphylococcal cassette chromosome mec identified in community-acquired methicillin-resistant Staphylococcus aureus strains. Antimicrob Agents Chemother 46:1147–1152
Maiden MCJ, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou JJ, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG (1998) Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95:3140–3145
Mazmanian SK, Hung IT, Schneewind O (2001) Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol Microbiol 40:1049–1057
McDevitt D, Foster TJ (1995) Variation in the size of the repeat region of the fibrinogen receptor (clumping factor) of Staphylococcus aureus strains. Microbiology 141:937–943
McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC (2003) Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol 41:5113–5120
McDowell P, Affas Z, Reynolds C, Holden MTG, Wood SJ, Saint S, Cockayne A, Hill PJ, Dodd CER, Bycroft BW, Chan WC, Williams P (2001) Structure, activity and evolution of the group I thiolactone peptide quorum-sensing system of Staphylococcus aureus. Mol Microbiol 41:503–512
Moore PCL, Lindsay JA (2001) Genetic variation among hospital isolates of methicillin-sensitive Staphylococcus aureus: evidence for horizontal transfer of virulence genes. J Clin Microbiol 39:2760–2767
Moore PCL, Lindsay JA (2002) Molecular characterisation of the dominant UK methicillin-resistant Staphylococcus aureus strains, EMRSA-15 and EMRSA16. J Med Microbiol 51:516–521
Moreau M, Richards JC, Fournier JM, Byrd RA, Karakawa WW, Vann WF (1990) Structure of the Type-5 capsular polysaccharide of Staphylococcus aureus. Carbohydr Res 201:285–297
Moxon ER, Rainey PB, Nowak MA, Lenski RE (1994) Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr Biol 4:24–33
Nakagawa I, Kurokawa K, Yamashita A, Nakata M, Tomiyasu Y, Okahashi N, Kawabata S, Yamazaki K, Shiba T, Yasunaga T, Hayashi H, Hattori M, Hamada S (2003) Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Res 13:1042–1055
Narita S, Kaneko J, Chiba J, Piemont Y, Jarraud S, Etienne J, Kamio Y (2001) Phage conversion of Panton–Valentine leukocidin in Staphylococcus aureus: molecular analysis of a PVL-converting phage, phi SLT. Gene 268:195–206
Nathwani D (2003) Impact of methicillin-resistant Staphylococcus aureus infections on key health economic outcomes: does reducing the length of hospital stay matter? J Antimicrob Chemother 51(Suppl 2):ii37–ii44
Novick RP (1963) Analysis by transduction of mutations affecting penicillinase formation in Staphylococcus aureus. J Gen Microbiol 33:121–136
Novick RP (1987) Plasmid incompatibility. Microbiol Rev 51:381–395
Novick RP, Projan SJ, Kornblum J, Ross HF, Ji G, Kreiswirth B, Vandenesch F, Moghazeh S (1995) The agr P2 operon: an autocatalytic sensory transduction system in Staphylococcus aureus. Mol Gen Genet 248:446–458
O’Riordan K, Lee JC (2004) Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev 17:218–234
Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MTG, Churcher CM, Bentley SD, Mungall KL, Cerdeno-Tarraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O’Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, Whitehead S, Barrell BG, Maskell DJ (2003) Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet 35:32–40
Paulsen IT, Firth N, Skurray RA (1997) Resistance to antimicrobial agents other than β-lactams. In: Crossley KB, Archer GL (eds) The staphylococci in human disease. Churchill Livingstone, New York, pp 175–212
Pearson H (2002) ‘Superbug’ hurdles key drug barrier. Nature 418:469
Pearson WR, Lipman DJ (1988) Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A 85:2444–2448
Phillips S, Novick RP (1979) Tn554-site-specific repressor-controlled transposon in Staphylococcus aureus. Nature 278:476–478
Preston GM, Haubold B, Rainey PB (1998) Bacterial genomics and adaptation to life on plants: implications for the evolution of pathogenicity and symbiosis. Curr Opin Microbiol 1:589–597
Projan SJ, Archer GL (1989) Mobilization of the relaxable Staphylococcus aureus plasmid pC221 by the conjugative plasmid pGO1 involves three pC221 loci. J Bacteriol 171:1841–1845
Projan SJ, Shlaes DM (2004) Antibacterial drug discovery: is it all downhill from here? Clin Microbiol Infect 10:18–22
Reed SB, Wesson CA, Liou LE, Trumble WR, Schlievert PM, Bohach GA, Bayles KW (2001) Molecular characterization of a novel Staphylococcus aureus serine protease operon. Infect Immun 69:1521–1527
Robinson DA, Enright MC (2003) Evolutionary models of the emergence of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 47:3926–3934
Roche FM, Massey R, Peacock SJ, Day NPJ, Visai L, Speziale P, Lam A, Pallen M, Foster TJ (2003) Characterization of novel LPXTG-containing proteins of Staphylococcus aureus identified from genome sequences. Microbiology 149:643–654
Rowland SJ, Dyke KGH (1989) Characterization of the staphylococcal beta-lactamase transposon Tn552. EMBO J 8:2761–2773
Ruzin A, Lindsay J, Novick RP (2001) Molecular genetics of SaPl1—a mobile pathogenicity island in Staphylococcus aureus. Mol Microbiol 41:365–377
Sau S, Bhasin N, Wann ER, Lee JC, Foster TJ, Lee CY (1997) The Staphylococcus aureus allelic genetic loci for serotype 5 and 8 capsule expression contain the type-specific genes flanked by common genes. Microbiology 143:2395–2405
Schneewind O, Model P, Fischetti VA (1992) Sorting of protein A to the staphylococcal cell wall. Cell 70:267–281
Smoot JC, Barbian KD, Van Gompel JJ, Smoot LM, Chaussee MS, Sylva GL, Sturdevant DE, Ricklefs SM, Porcella SF, Parkins LD, Beres SB, Campbell DS, Smith TM, Zhang Q, Kapur V, Daly JA, Veasy LG, Musser JM (2002) Genome sequence and comparative microarray analysis of serotype M18 group A Streptococcus strains associated with acute rheumatic fever outbreaks. Proc Natl Acad Sci U S A 99:4668–4673
Sumby P, Waldor MK (2003) Transcription of the toxin genes present within the staphylococcal phage phi Sa3ms is intimately linked with the phage’s life cycle. J Bacteriol 185:6841–6851
Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O’Connor KJ, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM (2005) Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pangenome”. Proc Natl Acad Sci U S A 102:13950–13955
Thakker M, Park JS, Carey V, Lee JC (1998) Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect Immun 66:5183–5189
Thomas WD, Archer GL (1989) Identification and cloning of the conjugative transfer region of Staphylococcus aureus plasmid pGO1. J Bacteriol 171:684–691
Thomas CM, Nielsen KM (2005) Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat Rev Microbiol 3:711–721
Tiemersma EW, Bronzwaer S, Lyytikainen O, Degener JE, Schrijnemakers P, Bruinsma N, Monen J, Witte W, Grundmann H (2004) Methicillin-resistant Staphylococcus aureus in Europe, 1999–2002. Emerg Infect Dis 10:1627–1634
Ubeda C, Maiques E, Knecht E, Lasa I, Novick RP, Penades JR (2005) Antibiotic-induced SOS response promotes horizontal dissemination of pathogenicity island-encoded virulence factors in staphylococci. Mol Microbiol 56:836–844
Ubelaker MH, Rosenblum ED (1978) Transduction of plasmid determinants in Staphylococcus aureus and Escherichia coli. J Bacteriol 133:699–707
van der Woude MW, Baumler AJ (2004) Phase and antigenic variation in bacteria. Clin Microbiol Rev 17:581–611, table of contents Functional and Integrative Genomics Page 40 of 47
van Duijkeren E, Box ATA, Heck M, Wannet WJB, Fluit AC (2004) Methicillin-resistant staphylococci isolated from animals. Vet Microbiol 103:91–97
Vautor E, Abadie G, Guibert JM, Chevalier N, Pepin M (2005) Nasal carriage of Staphylococcus aureus in dairy sheep. Vet Microbiol 106:235–239
Watts A, Ke DB, Wang Q, Pillay A, Nicholson-Weller A, Lee JC (2005) Staphylococcus aureus strains that express serotype 5 or serotype 8 capsular polysaccharides differ in virulence. Infect Immun 73:3502–3511
Weigel LM, Clewell DB, Gill SR, Clark NC, McDougal LK, Flannagan SE, Kolonay JF, Shetty J, Killgore GE, Tenover FC (2003) Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 302:1569–1571
Williams RJ, Ward JM, Henderson B, Poole S, O’Hara BP, Wilson M, Nair SP (2000) Identification of a novel gene cluster encoding staphylococcal exotoxin-like proteins: characterization of the prototypic gene and its protein product, SET1. Infect Immun 68:4407–4415
Yamaguchi T, Hayashi T, Takami H, Nakasone K, Ohnishi M, Nakayama K, Yamada S, Komatsuzawa H, Sugai M (2000) Phage conversion of exfoliative toxin A production in Staphylococcus aureus. Mol Microbiol 38:694–705
Zetola N, Francis JS, Nuermberger EL, Bishai WR (2005) Community-acquired methicillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect Dis 5:275–286
Ziebuhr W, Ohlsen K, Karch H, Korhonen T, Hacker J (1999) Evolution of bacterial pathogenesis. Cell Mol Life Sci 56:719–728
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lindsay, J.A., Holden, M.T.G. Understanding the rise of the superbug: investigation of the evolution and genomic variation of Staphylococcus aureus . Funct Integr Genomics 6, 186–201 (2006). https://doi.org/10.1007/s10142-005-0019-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-005-0019-7