
Abstract
Molluscan shell color polymorphism is important in genetic breeding, while the molecular information mechanism for shell coloring is unclear. Here, high-throughput RNA sequencing was used to compare expression profiles of coding and non-coding RNAs (ncRNAs) from Pacific oyster Crassostrea gigas with orange and black shell, which were from an F2 family constructed by crossing an orange shell male with a black shell female. First, 458, 13, and 8 differentially expressed genes (DEGs), lncRNAs (DELs), and miRNAs (DEMs) were identified, respectively. Functional analysis suggested that the DEGs were significantly enriched in 9 pathways including tyrosine metabolism and oxidative phosphorylation pathways. Several genes related to melanin synthesis and biomineralization expressed higher whereas genes associated with carotenoid pigmentation or metabolism expressed lower in orange shell oyster. Then, based on the ncRNA analysis, 163 and 20 genes were targeted by 13 and 8 differentially expressed lncRNAs (DELs) and miRNAs (DEMs), severally. Potential DELs-DEMs-DEGs interactions were also examined. Seven DEMs-DEGs pairs were detected, in which tyrosinase-like protein 1 was targeted by lgi-miR-133-3p and lgi-miR-252a and cytochrome P450 was targeted by dme-miRNA-1-3p. These results revealed that melanin synthesis-related genes and miRNAs-mRNA interactions functioned on orange shell coloration, which shed light on the molecular regulation of shell coloration in marine shellfish.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Shell coloration, being used as highly tractable makers for selective breeding, has long fascinated breeders and scientists (Alfnes et al. 2006). Nowadays, many phenotypic varieties have been obtained in shellfish (Hu et al. 2019; Nie et al. 2020; Wang et al. 2017). Additional studies have been identified that color polymorphism is inheritable and may be tightly regulated by a set of genes (Evans et al. 2009; Kobayashi et al. 2004; Winkler et al. 2001). However, compared with plants, vertebrates, and certain insects, the molecule mechanism leading to shell coloration has not been completely elucidated for shellfish (Mann and Jackson 2014), which is a serious gap in our knowledge of how color has evolved in the natural world (Williams 2017). Hence, identifying and understanding the genetic basis underlying shell color traits have become a key goal of genetics.
Colorful oyster shell is mainly because of the presence of biological pigments, and shell color formation closely depends on the creation, deposition, and transport of pigments. Melanin, carotenoids, and tetrapyrroles are the main pigments in molluscan shells (Williams 2017). Eumelanin, one kind of melanin, is responsible for black or brown, and pheomelanin, the other kind of melanin, can be yellow to red. In addition, carotenoids are yellow, red, and orange pigments (Orteu and Jiggins 2020). Due to the ongoing advances in the high-throughput sequencing, extensive molluscan mantle transcriptome data have been analyzed; these studies identified common genes and pathways related to the pigment synthesis, such as tyrosinase and cytochrome P450 gene as well as tyrosine metabolism pathway, etc., suggesting participation in the regulation of shell pigmentation (Bai et al. 2013; Ding et al. 2015; Feng et al. 2015; Hu et al. 2019; Sun et al. 2016). In addition, shell color is also relevant to shell construction (Williams 2017). Transcriptomic and proteomic study has suggested that shell pigmentation might be connected to special shell structure and biomineralization-related genes, such as glycine-rich protein, matrix proteins, calmodulins, and others (Lemer et al. 2015; Xu et al. 2019). Though a number of studies have been conducted on shell color formation, the precise genes and molecular pathways that underlie the regulation of shell pigmentation remain largely unknown.
Indeed, transcriptional and post-transcriptional regulation of gene expression is regulated by non-coding RNAs (ncRNAs), comprising different types of small non-coding RNAs (sRNAs) and long non-coding RNAs (lncRNAs) (Zhang et al. 2021). So far, it has been verified that ncRNAs were involved in a variety of biological processes in mollusks, including development, biomineralization, and innate immune response (Huang et al. 2018; Zheng et al. 2016, 2019; Zhu et al. 2020). In terms of skin color formation, studies revealed that ncRNAs acted important roles in fish skin color differentiation (Luo et al. 2019; Tian et al. 2018; Wang et al. 2018). Nevertheless, to our knowledge, only a little report focused on the roles of ncRNAs on pigmentation in mollusks (Chen et al. 2019; Feng et al. 2018, 2020).
The Pacific oyster, Crassostrea gigas, is one of the most economically important marine bivalve species. In our breeding program, a novel orange shell color variant of C. gigas was obtained (Han et al. 2019), and the orange shell color is a recessive trait compared to black and white (Han and Li 2020). Recently, an F2 family was constructed by crossing an orange shell male with a black shell female (Han and Li 2020), which provides an ideal population for investigating and gaining a better understanding of the shell color formation mechanism in C. gigas. Herein, whole transcriptome sequencing was performed in the orange and black shell oysters from the same F2 family to analyze the transcriptional patterns for mRNAs, lncRNAs, and miRNAs that may function to regulate shell color.
Materials and Method
Sampling
The orange and black shell color oysters used in this study were taken from previous breeding process. In July 2017, a black female and an orange male were crossed to produce the first-generation (F1) family. In June 2018, three females and three males were randomly selected from the F1 family and mated to construct three second-generation (F2) families (Han and Li 2020). One year later, the orange and black shell strains were randomly sampled from three F2 families. Left mantles of 9 individuals of each strain were immediately snap frozen in liquid nitrogen individually and stored at − 80 °C until use. Three mantles from each strain of oysters were pooled for RNA isolation and three biological replicates were performed for one pool. One pool mantle from orange shell color and black shell color was denoted as OM and BM, respectively.
RNA Isolation, Library Preparation for LncRNA, and Small RNA and Sequencing
Total RNA was isolated with Trizol reagent (Invitrogen, CA, USA) following the manufacturer’s protocol. Then, RNA purity and integrity were checked using NanoPhotometer® spectrophotometer (IMPLEN, CA, USA) and Agilent Bioanalyzer 2100 system with RNA 6000 Nano Kit (Agilent Technologies, CA, USA). RNA concentration was measured using Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA). A total amount of 3 µg RNA per sample was used as input material for the RNA sample preparations. LncRNA sequencing libraries and small RNA sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit and NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (NEB, USA) following manufacturer’s recommendations, respectively. After the cDNA synthesis and PCR amplification, the sequencing of each cDNA library was performed on Illumina novaseq 6000 platform at Novogene Bioinformatics Technology Co., Ltd. (Beijing, China) and raw reads were generated. All RNA-seq raw data were deposited into the Sequence Read Archive (SRA) database from NCBI (https://www.ncbi.nlm.nih.gov/sra). The BioProject accession number was PRJNA694837.
LncRNA Sequencing Analysis
Quality control and reads statistics were determined by Fast QC (Andrews 2010). Clean reads were produced by removing reads containing adapter, uncertain ‘N’ with the ration of ‘N’ > 10%, and low-quality reads from raw reads. For each RNA-seq sample, the high-quality clean reads were mapped to the newest reference C. gigas genome (RefSeq: GCF_902806645.1) using Hisat 2 (Kim et al. 2015). The mappable reads were assembled by Stringtie (2.1.1). Transcripts from all samples were then merged together with Stringtie merge mode to build a consensus set of transcripts across samples. Transcript abundances were estimated and read coverages were generated using Stringtie.
The pipeline of novel lncRNAs prediction was adapted with few modifications from Azlan et al. (2019) and sequencing company; five filters were used to generate lncRNA: (1) transcripts shorter than 200 bp and with FPKM and TPM less than 1 were eliminated; (2) transcripts with class code ‘i’, ‘u’, ‘x’ subsets were retained; (3) transcripts belong to coding genes were excluded using TransDecoder; (4) protein coding potency of transcripts were calculated by Coding Potential Assessment Tool (CPAT) (coding prob > 0.3) (Wang et al. 2013), Coding Non-Coding Index (CNCI) (score > 0) (Sun et al. 2013) and BLAST against Pfam and Swissprot database (E-value < 10–6), transcripts identified with coding potential in any of the three methods were filtered out; and (5) transcripts without strand information were removed.
Small RNA Sequencing Analysis
The small RNA sequencing raw reads with the content of unknown base N ≥ 10% and low quality were removed. Reads without 3′ adaptor sequence and inserted fragment were removed. Sequences shorter than 18 nucleotides or longer than 35 nucleotides were removed and clean reads were obtained. The high-quality clean reads were mapped to the newest reference C. gigas genome (RefSeq: GCF_902806645.1) using bowtie 2 (Langmeada and Salzberg 2012). The matched clean reads were aligned with Rfam database to remove rRNA, scRNA, snoRNA, snRNA, and tRNA, then aligned with repeat sequences predicted by RepeatMasker (Tarailo-Graovac and Chen 2009). The filtered reads were searched against the reference genome to remove the reads mapped in exons. Subsequently, the remained reads were blasted to invertebrate miRNAs in miRbase (http:// www.mirbase.org/) to identify known miRNA in C. gigas. Invertebrate including Strongylocentrotus purpuratus, Caenorhabditis elegans, Lottia gigantea, Nematostella vectensis, and Drosophila melanogaster were used in this study. The remaining un-annotated reads were predicted to be novel miRNA by Mireap (Chen et al. 2009). The outputs were then manually checked according to the requisite criteria for miRNA annotation proposed in recent reports.
Differential Expression and Function Enrichment Analysis
The quantification of lncRNAs and mRNAs in each sample was calculated by Stringtie (Pertea et al. 2015). Identification of differentially expressed transcripts between OM and BM group was performed using DESeq2 v1.26.0 (Love et al. 2014). Adjusted p value < 0.05 and |fold change|> 2 were considered as significantly differential expression of mRNAs (DEGs) and lncRNAs (DELs). The expression levels of miRNAs were normalized with TPM algorithm (Zhou et al. 2010). Adjusted p value < 0.05 was considered as significantly differently expressed miRNAs (DEMs). Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genome (KEGG) pathway enrichment analysis of DEGs were conducted using the TBtools (Chen et al. 2020).
Association Analysis Between mRNAs and ncRNAs
The cis role of lncRNAs was to act on their neighboring target genes (Ørom et al. 2010). To reduce false positives, the genes located within the 10 kb distance of the lncRNAs were selected as potential target genes using Bedtools (2.29.2). Pearson’s correlation coefficients of gene expression were calculated for each lncRNAs-mRNAs pair. Then, the pairs of miRNAs-lncRNAs or miRNAs-mRNAs were predicted using miRanda; an alignment score N = 150 and minimum free energy of − 7 kcal/mol were set as the threshold. The pairwise correlations of miRNAs-lncRNAs or miRNAs-mRNAs were evaluated using the Pearson correlation coefficient, and every pair was selected by RNA pairs of p < 0; then, the predicted miRNAs-lncRNAs pairs were integrated with the miRNAs-mRNAs pairs by the shared miRNAs. Finally, the interaction network was visually displayed using Cytoscape v3.8.0.
Quantitative Real-Time PCR Validation of Differentially Expressed Transcripts
To validate the expression results of transcriptome data, a number of DEGs, DELs, and DEMs were randomly selected for real-time qPCR. For qPCR of mRNAs and lncRNAs, the total RNA was reverse transcribed into cDNA using PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, China), according to the manufacturer’s instructions. The internal reference gene was ef1α (Du et al. 2013). For qPCR of miRNAs, the total RNA was reverse transcribed into cDNA using miRNA 1st strand cDNA synthesis kit (by stem-loop) (Vazyme, China). The reverse primer was universal and the forward primers were unique. U6 snRNA was used as the internal reference for normalization (Chen et al. 2019). Primers used for qPCR validation are listed in Table 1. The qPCR was carried out in 20 μL using QuantiNova™ SYBR® Green PCR Kit (QIAGEN, Germany) for mRNAs and lncRNAs and miRNA Universal SYBR® PCR Master Mix (Vazyme, China) for miRNAs on LightCycler 480 real-time PCR instrument (Roche Diagnostics, Burgess Hill, Switzerland). Each qPCR reaction was performed in triplicate, and relative expression was calculated using 2−ΔΔCT methods (Livak and Schmittgen 2001).
Results
Overview of RNA Sequencing and Identification of mRNAs, lncRNAs, and miRNAs
In total, 98.01% of raw reads were clean reads in BM group and 97.66% of raw reads were clean reads in OM group. Six cDNA libraries were constructed from BM and OM groups, yielding 15.38 Gb and 14.75 Gb of clean data with Q20 values of 98.12% for BM group and 98.21% for OM group. A total of 77.26% and 81.07% of clean reads from BM and OM groups were aligned to the Pacific oyster genome (Table S1). Furthermore, a single merged transcriptome containing 94,472 transcripts and 40,661 gene loci was obtained. The high-quality clean reads were subjected to an optimized method to identify mRNAs and lncRNAs. A total of 31,950 mRNAs were identified. And the final number of lncRNAs was 955, including 292 intronic, 641 intergenic, and 22 antisense lncRNAs. Comparison results of the genomic characterizations about lncRNAs and mRNAs showed that the length distributions of lncRNAs and mRNAs differed, and the peak of lncRNAs was at about 9 while the other at about 11 (Fig. 1a). In addition, less GC content and lower overall expressed levels were found in lncRNAs than mRNAs (Fig. 1b, c). In terms of minimum free energy, lncRNAs were slightly more than mRNAs (Fig. 1d).

Characteristics of mRNA, lncRNA, and miRNA. a–c Comparison of genomic architecture and expression level of lncRNAs and mRNAs. a Distribution of lengths of lncRNAs and mRNAs. b GC contents of lncRNAs and mRNAs. c Expression level of lncRNAs and mRNAs, calculated as log10(FPKM + 1). d Minimum free energy (MFE kcal/mol) of lncRNAs and mRNAs
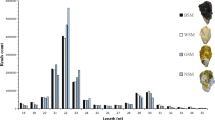
The small RNA libraries of BM and OM groups were determined and analyzed by Illumina deep sequencing. A total of 48,961,810 and 59,170,092 clean reads were obtained from the BM and OM libraries, respectively. After a series of filters, 42,981,200 (87.79%) and 52,838,341 (89.30%) clean reads were retrieved for further analysis. Moreover, 90.59% clean reads were mapped to the Pacific oyster genome (Table S2). The length of small RNAs (sRNAs) ranged from 18 to 35 nt, with 22 nt sRNAs representing the most frequent lengths (Fig. 2). In order to annotate the different classes of sRNAs, clean reads were compared with miRBase, ncRNA, exon, and repeat-associated RNA databases (Table S2). Finally, a total of 89 miRNAs (77 conserved and 12 novel miRNAs) were identified; among these, 47 known miRNAs were identified based on miRNA database of L. gigantea in miRbase. The high conservation of the known 30 detected miRNAs with other species’ miRNAs in miRbase (Table S3).

The length distribution of small RNAs
Function Enrichment Analysis and Potential DEGs Related to the Shell Coloring Detection
In total, 458 DEGs including 238 up-regulated genes and 220 down-regulated genes were identified in OM and BM comparison (Fig. 3a). GO enrichment analyses revealed that 458 DEGs were enriched in 14, 13, 3 GO terms in biological process, cellular components, and molecular function, respectively. Within the three process categories, gene expression, ribosome, and structural constituent of ribosome were the most represented, respectively (Fig. 3b). KEGG pathway analyses showed that DEGs were significantly enriched in 9 KEGG pathways, in which genetic information processing was the most enriched. Moreover, pathways related to melanogenesis and ATP synthesis like tyrosine metabolism, oxidative phosphorylation, and energy metabolism pathway were significantly enriched (Fig. 3c).

Identification of enriched analysis of DEGs. Volcano plot of differentially expressed genes (DEGs) in comparison of OM and BM. X-axis exhibited change in fold between samples and Y-axis shows significance of DEGs. Red (up-regulation) and blue (down-regulation) dots indicated significantly different expression (q-value < 0.05, |log2 (fold change) |> 1), respectively, and dots in gray color showed no significant differences. b Gene ontology classification of 458 DEGs in comparisons between OM and BM. Blue, orange, and green represent biological process, cellular component, and molecular function, respectively. c Enriched Kyoto encyclopedia of genes and genomes (KEGG) pathways of 458 DEGs comparisons between OM and BM
In order to investigate the molecular mechanisms of orange shell formation in C. gigas, some DEGs related to shell color formation were explored (Table 2). Genes involved in melanin synthesis like tyr1, sfrp3, calm1 were differentially up-regulated in OM group compared with BM group. Most genes related to the carotenoid pigmentation were significantly down-regulated in OM group. With respect to CYP450 in OM group, gene expressions of two subtypes were increased; other subtypes were decreased. Cytochrome c oxidase is related to ATP synthesis. In this study, transcriptional level of cox19 and coa7 was both higher in OM group than BM group. Furthermore, many genes related to biomineralization, such as matrix proteins, calmodulins, and collagen alpha chain, were significantly up-regulated in OM group.
Expression Profiling of ncRNAs and Their Target Gene Prediction
The expression of ncRNAs regulates the expression of the protein-coding genes. Here, a total of 13 DELs were identified, 4 lncRNAs exhibited higher expression level in OM group than that in BM group, while 9 lncRNAs showed lower expression level in OM group. Additionally, 8 DEMs were identified. Compared with BM group, 3 miRNAs were up-regulated in OM group, including lgi-miR-31, dme-miR-1-3p, and lgi-miR-279, while 5 miRNAs were down-regulated (Table S4). These DELs and DEMs reflected their specific functions and related biological mechanisms in the mantle of orange shell oyster.
The potential cis targets of lncRNAs were predicted to investigate the functions of 13 DELs, and 13 DELs were corresponded to 163 protein-coding genes (Fig. 4a, Table S5). In terms of target gene prediction in miRNAs, a total of 20 target genes were predicted for 8 DEMs (Fig. 4b). Except lgi-miR-133-3p, other DEMs appear to target multiple genes. Moreover, some genes were regulated by more than one miRNA (Fig. 4b). To reveal the global regulatory network of mRNAs and ncRNAs about shell pigmentation, a regulation network was constructed using DEMs-DEGs pairs and DEMs-DELs pairs, every pair was selected by RNA pairs of p < 0. In total, 7 DEMs-DEGs pairs were detected, 5 DEGs were predicted as targets of 5 DEMs (Fig. 4c). As follows, tyr1 gene (LOC105331192) was negatively regulated by lgi-miR-133-3p and lgi-miR-252a, timp3 gene (LOC105346390) was negatively regulated by dme-miR-981-3p, CYP 4F2-like isoform X1 gene (LOC105335484) was negatively regulated by dme-miRNA-1-3p, suggesting that these DEMs likely played a key role in the shell color variation process.

Regulation analysis in mRNAs, lncRNAs, and miRNAs. Orange: up-regulation, green: down-regulation, ellipse: mRNAs, diamond: lncRNAs, V type: miRNAs. a Cis-regulations between differential expression lncRNAs (DELs) and mRNAs. b Regulations between differential expression miRNAs (DEMs) and mRNAs. c Regulations between DEMs and differential expression mRNAs (DEGs)
The qPCR Validation
To validate the RNA-seq results, eight DEGs, 2 DELs, and 4 DEMs were randomly selected to confirm the reliability of the sequencing technology. The expressions of 6 DEGs related to shell color and biomineralization were in line with the expression results of RNA-seq, except that fib and calm1 genes were not significantly changed (Fig. 5). Two candidate up-regulated genes, rpl24 and tyr1, were predicted to be the target genes of down-regulated miRNAs dme-miR-981-3p, lgi-miR-252a, and lgi-miR-133-3p, respectively. Above 3 down-regulated miRNAs and 1 up-regulated miRNA, i.e., lgi-miR-279, were selected to validate; the results from qPCR matched those from RNA-Seq analysis (Fig. 5). Two lncRNAs also were assessed by qPCR; the results showed that expression levels of LOC109619958 and LOC105340081 were significantly different between OM and BM group, as described in RNA-Seq analysis (Fig. 5).

Quantitative PCR validation of DEGs, DELs, and DEMs. Results were shown as means ± standard deviation of triplicate measurements. “*” and “#” indicate p < 0.05. Gray and white bar stands for qPCR and RNA-seq results, respectively. DEGs: rpl24, 60S ribosomal protein L24, tyr1, tyrosinase-like protein 1, rarg, retinoic acid receptor gamma isoform X2, ebna1bp2, probable rRNA-processing protein EBP2, fib, nacrein-like protein isoform X1, CYP7A1, cytochrome P450 7A1, calm1, neo-calmodulin-like, noth1, neurogenic locus notch homolog protein 1-like; DEL: LOC109619958 and LOC105340081. DEMs: lgi-miR-252a, lgi-miR-279, dme-miR-981-3p, and lgi-miR-133-3p
Discussion
Reliable and abundant lncRNA database and small RNA database in mantle of C. gigas with orange and black shell color were constructed using high-throughput sequencing. In total, 955 putative lncRNAs were characterized; these lncRNAs shared many common characteristics among species, including shorter transcript length, lower GC content, and lower expression as compared to known protein-coding transcripts (Cabili et al. 2011). These features were also observed in this study, indicating that lncRNAs identified were reliable. The sRNA libraries constructed displayed a similar read length distribution of 21–23 nt sRNAs; the most frequent length of sRNA sequenced was 22 nt, which is equivalent in size to typical Dicer-derived products (Starega-Roslan et al. 2015), suggesting the conservation of miRNAs. A total of 89 miRNAs (77 conserved and 12 novel miRNAs) were identified, among these, 47 known miRNAs identified based on miRNA database of L. gigantea, which is consistent with the previous results in C. gigas (Feng et al. 2020). The high conservation of the known 30 detected miRNAs with other species’ miRNAs in miRBase 21.0 suggested the conservation of miRNA sequences in the evolution process of different species (Wheeler et al. 2009).
The expression patterns of mRNAs and ncRNAs were further investigated to gain insight into their possible roles in orange shell color formation in C. gigas. A total of 458 DEGs were identified in OM and BM comparison; enrichment study suggested that DEGs significantly enriched in the tyrosine metabolism related to melanogenesis and may play a vital role in distinguishing orange and black shell oyster. DEGs were also significantly enriched in “oxidative phosphorylation” related to ATP synthesis and may be involved in pigmentation. Real-time qPCR experiments were carried out to validate the RNA-seq data; results indicated that the RNA-seq was a reliable reference for expression profiling study.
In this study, many pigmentation-related genes were screened in differentially expressed gene analysis. Tyrosinases are essential for melanogenesis (Aguilera et al. 2014). The upregulation of tyrosinase gene has been reported in the bivalves Patinopecten yessoensis and QN Orange scallops, showing contrasting pigmentation phenotypes (Ding et al. 2015; Song and Wang 2019). In this study, tyr1 gene exhibited higher expression level in the mantle of orange shell phenotype than black shell phenotype, suggesting that tyrosinase had an influence on orange coloration compared with black coloration. Moreover, sfrp3 is an essential factor associated with wnt signaling pathway, which is involved in the maintenance of melanocyte and keratinocyte homeostasis (Ekstro et al. 2011). Here, sfrp3 gene expression was elevated in orange shell phenotype, which accelerated the melanin production. Furthermore, it has been found that CYP450 also implicated in the melanin biosynthesis by catalyzing retinoate into retinoic acid (Yu et al. 2017). And Song and Wang (2019) suggested that CYP450 might affect orange coloration through the accumulation of melanin in QN orange scallops. Surprisingly, several genes involved in melanin synthesis were expressed at higher levels in orange shell oyster compared with the black strain, which indicated that melanin production might play an important role in orange shell coloration. Melanins are known to exit in two forms in mollusks: eumelanin, a brown or black pigment; and pheomelanin, a red, yellow, or brown pigment (Williams 2017). Hines et al. (2017) pointed out that black and red setae differ primarily in relative amount of melanin, with black setae containing some combination of both eumelanin and pheomelanin and red containing exclusively pheomelanin. Namely, black and red setae in insects depended mainly on relative amount of eumelanin and pheomelanin. In C. gigas, the contents of eumelanin and pheomelanin of shell in orange and black shell have been studied in our research group; results showed that ratios of red pheomelanin to dark eumelanin were higher in orange shell than the black (unpublished data). We are therefore speculated that high expressions of tyr1, sfrp3, and CYP450 genes in orange shell might contribute to the pheomelanin synthesis. On the other hand, it has been reported that red carotenoid coloration was controlled by a CYP450 gene cluster in the Zebra Finch (Mundy et al. 2016). In this study, different members of CYP450 gene family had diverse differential expressions in orange shell phenotype compared with the black shell phenotype, thus the possibility that CYP450 affected orange coloration through the accumulation of different pigments including pheomelanin and carotenoid in C. gigas could not be excluded. Additionally, RBP4, RAR, and LDLR had known function related to the biosynthesis, accumulation, and metabolism of carotenoids. RBP4, RAR, and LDLR genes were expressed at lower levels in orange shell phenotype than the black. Gene expression results were consistent with the relative contents of carotenoids in mantles of C. gigas with orange and black shell color (Fig. S1).
Shell color is mainly determined by the presence of pigments produced by the mantle, but the calcium carbonate and microstructure of the shell may also contribute to coloration (Saenko and Schilthuizen 2021). The determination of crystal type (aragonite or calcite) and the formation of microstructures such as the prismatic and the nacreous layers are thought to be regulated by organic matrix proteins (Belcher et al. 1996; Falini et al. 1996). Proteins produced in the mantle are secreted into the extrapallial space, where calcium carbonate (CaCO3) crystallizes to build unusual microstructures (Yano et al. 2006). The mechanism of this process is unknown, but may involve interactions of the matrix proteins and inorganic ions present in the extrapallial space, leading to crystallization of CaCO3 and morphogenesis of the species-specific appearance of the shell (Yano et al. 2006). Lemer et al. (2015) pointed out that the origin shell color was induced by shell matrix protein expressed in the nacre and calcitic layer of pearl oysters (Pinctada margaritifera). In our study, the expression levels of genes related to biomineralization, such as matrix proteins, calmodulins, and collagen alpha chain, were higher in orange shell oyster, suggesting that matrix proteins and calmodulin genes were most likely involved in the establishment of special shell microstructure in orange phenotype that differed from black phenotype. In addition, multiple transcriptome analysis in pearl oyster also detected differential expression of many biomineralization-related genes and suggested that shell pigmentation required essential biomineralization genes (Auffret et al. 2020; Xu et al. 2019); however, this connection contributing to the pigmentation difference between orange and black shell in the Pacific oyster needed to be further studied.
The role of miRNA is to inhibit protein synthesis by partially or completely binding to the 3′ untranslated region of mRNA, thus regulating gene expression (Vidigal and Ventura 2015). Although a large number of miRNAs have been identified in melanocyte biology of mammals, amount of miRNA in oyster pigmentation has rarely been studied (Zhang et al. 2021). In this study, a total of 20 target genes were predicted for 8 DEMs (Fig. 4b). Multiple genes were targeted by a single miRNA, suggesting a multiple regulatory function for these miRNAs in multiple signal pathways. Within these pairs, 5 DEGs were predicted as targets of 5 DEMs, of which, tyr1 gene, acting as a key role in melanin synthesis, was negatively regulated by lgi-miR-133-3p and lgi-miR-252a. CYP 450 gene, implicating in the production of melanin and carotenoid, was negatively regulated by dme-miR-1-3p. MiR-133b and its target gene, SLC associated with pigmentation, and melanogenesis have been detected to influence muscle melanogenesis in Muchuan black-boned chickens (Yu et al. 2020). So, it was likely that lgi-miR-133-3p and its target gene (tyr1) may play an important role in shell pigmentation by influencing melanin synthesis in C. gigas. The regulation of tyr gene expression in different carp has been reported to be affected by different miRNAs (Tian et al. 2018; Yin et al. 2020), suggesting that the regulation of tyr gene expression level may be regulated by diverse miRNAs even in the same species. Although the function on pigmentation of lgi-miR-252a and dme-miR-1-3p has not been reported in animals, in current studies, lgi-miR-252a and dme-miR-1-3p might be major miRNAs acting on shell coloration via regulating DEG-associated pigment synthesis. The functional roles of such DEMs are the focus of future investigation. Indeed, compared with the previous study in four shell color strains of C. gigas (white, golden, black, and partially pigmented) (Feng et al. 2020), novel functional miRNAs being possible for regulating shell pigmentation were explored in orange and black shell color of C. gigas. Hence, the study of small RNA sequencing provides a new clue about the pigmentation function of miRNAs in C. gigas and it is necessary to uncover more miRNAs by incessant studies.
Conclusion
In this study, the whole transcriptome sequencing of mantles was performed in C. gigas with orange and black shell. The expressions of DEGs involved in melanogenesis and biomineralization were up-regulated in orange shell oyster, which implied that orange shell coloration in C. gigas may be controlled by genes modulating accumulation of melanin and biomineralization processes. Additionally, three DEMs (lgi-miR-133-3p, lgi-miR-252a, and dme-miRNA-1-3p) and their target genes related to pigment synthesis were found, and the roles of these DEMs in pigment synthesis needed to be further studied. This study provided valuable information for understanding the miRNA-mediated regulatory mechanisms underlying shell coloration in marine shellfish.
Abbreviations
- tyr1 :
-
Tyrosinase-like protein 1
- sfrp3 :
-
Secreted frizzled-related protein 3
- calm1 :
-
Neo-calmodulin-like
- CYP450 :
-
Cytochrome P450
- cox19 :
-
Cytochrome c oxidase assembly protein COX19-like
- coa7 :
-
Cytochrome c oxidase assembly factor 7-like
- timp3 :
-
Metalloproteinase inhibitor 3-like
- fib :
-
Nacrein-like protein isoform X1
- rpl24 :
-
60S ribosomal protein L24
- RBP4 :
-
Retinol-binding protein 4
- RAR :
-
Retinoic acid receptor
- LDLR :
-
Low-density lipoprotein receptor
- SLC :
-
Solute carrier
References
Aguilera F, McDougall C, Degnan BM (2014) Evolution of the tyrosinase gene family in bivalve molluscs: independent expansion of the mantle gene repertoire. Acta Biomater 10:3855–3865
Alfnes F, Guttormsen AG, Steine G, Kolstad K (2006) Consumers’ willingness to pay for the color of salmon: a choice experiment with real economic incentives. Am J Agric Econ 88:1050–1061
Andrews S (2010) FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. 21 June 2020
Auffret P, Le Luyer J, Sham Koua M, Quillien V, Ky CL (2020) Tracing key genes associated with the Pinctada margaritifera albino phenotype from juvenile to cultured pearl harvest stages using multiple whole transcriptome sequencing. BMC Genom 21:662
Azlan A, Obeidat SM, Yunus MA, Azzam M (2019) Systematic identification and characterization of Aedes aegypti long noncoding RNAs (lncRNAs). Sci Rep 9:12147
Bai ZY, Zheng HF, Lin JY, Wang GL, Li GL (2013) Comparative analysis of the transcriptome in tissues secreting purple and white nacre in the pearl mussel Hyriopsis cumingii. PLoS One 8:e53617
Belcher AM, Wu XH, Christensen RJ, Hansma PK, Stucky GD, Morse DE (1996) Control of crystal phase switching and orientation by soluble mollusc-shell proteins. Nature 381:56–58
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinnl JL (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25:1915–1927
Chen CJ, Chen H, Zhang Y, Thomas H, Frank MH, He YH, Xia R (2020) TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant 13:1194–1202
Chen X, Li QB, Wang J, Guo X, Jiang XR, Ren ZJ, Weng CY, Sun GX, Wang XQ, Liu YP, Ma LJ, Chen JY, Wang J, Zen K, Zhang JF, Zhang CY (2009) Identification and characterization of novel amphioxus microRNAs by Solexa sequencing. Genome Biol 10:R78
Chen XJ, Bai ZY, Li JL (2019) The mantle exosome and microRNAs of Hyriopsis cumingii involved in nacre color formation. Mar Biotechnol 21:634–642
Ding J, Zhao L, Chang YQ, Zhao WM, Du ZL, Hao ZL (2015) Transcriptome sequencing and characterization of Japanese Scallop Patinopecten yessoensis from different shell color lines. PLoS One 10:e0116406
Du Y, Zhang LL, Xu F, Huang BY, Zhang GF, Li L (2013) Validation of housekeeping genes as internal controls for studying gene expression during Pacific oyster (Crassostrea gigas) development by quantitative real-time PCR. Fish Shellfish Immunol 34:939–945
Ekstro EJ, Sherwood V, Andersson T (2011) Methylation and loss of secreted frizzled-related protein 3 enhances melanoma cell migration and invasion. PLoS One 6:e18674
Evans S, Camara MD, Langdon CJ (2009) Heritability of shell pigmentation in the Pacific oyster, Crassostrea gigas. Aquaculture 286:211–216
Falini G, Albeck S, Weiner S, Addadi L (1996) Control of aragonite or calcite polymorphism by mollusk shell macromolecules. Science 271:67–69
Feng DD, Li Q, Yu H, Zhao XL, Kong LF (2015) Comparative transcriptome analysis of the Pacific Oyster Crassostrea gigas characterized by shell colors: identification of genetic bases potentially involved in pigmentation. PLoS One 10:e0145257
Feng DD, Li Q, Yu H, Kong LF, Du SJ (2018) Transcriptional profiling of long non-coding RNAs in mantle of Crassostrea gigas and their association with shell pigmentation. Sci Rep 8:1436
Feng DD, Li Q, Yu H, Liu SK, Kong LF, Du SJ (2020) Integrated analysis of microRNA and mRNA expression profiles in Crassostrea gigas to reveal functional miRNA and miRNA-targets regulating shell pigmentation. Sci Rep 10:20238
Han ZQ, Li Q (2020) Mendelian inheritance of orange shell color in the Pacific oyster Crassostrea gigas. Aquaculture 516:734616
Han ZQ, Li Q, Liu SK, Yu H, Kong LF (2019) Genetic variability of an orange-shell line of the Pacific oyster Crassostrea gigas during artificial selection inferred from microsatellites and mitochondrial COI sequences. Aquaculture 508:159–166
Hines HM, Witkowski P, Wilson JS, Wakamatsu K (2017) Melanic variation underlies aposematic color variation in two hymenopteran mimicry systems. PLoS One 12:1-17
Hu Z, Song H, Yang MJ, Yu ZL, Zhou C, Wang XL, Zhang T (2019) Transcriptome analysis of shell color-related genes in the hard clam Mercenaria mercenaria. Comp Biochem Physiol Part D Genomics Proteomics 31:100598
Huang XD, Dai JG, Lin KT, Liu M, Ruan HT, Zhang H, Liu WG, He MX, Zhao M (2018) Regulation of IL-17 by lncRNA of IRF-2 in the pearl oyster. Fish Shellfish Immunol 81:108–112
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357-U121
Kobayashi T, Kawahara I, Hasekura O, Kijima A (2004) Genetic control of bluish shell color variation in the Pacific abalone, Haliotis discus hannai. J Shellfish Res 23:1153–1156
Langmeada B, Salzberg SL (2012) Fast gapped-read alignment with bowtie 2. Nat Methods 9:357-U54
Lemer S, Saulnier D, Gueguen Y, Planes S (2015) Identification of genes associated with shell color in the black-lipped pearl oyster, Pinctada margaritifera. BMC Genom 16:568–582
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(T) (-Delta Delta C) method. Methods 25:402–408
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Luo MK, Wang LM, Yin HR, Zhu WB, Fu JJ, Dong ZJ (2019) Integrated analysis of long non-coding RNA and mRNA expression in different colored skin of koi carp. BMC Genet 20:515
Mann K, Jackson DJ (2014) Characterization of the pigmented shellforming proteome of the common grove snail Cepaea nemoralis. BMC Genom 15:249
Mundy NI, Stapley J, Bennison C, Tucker R, Twyman HL, Kim KW, Burke T, Birkhead TR, Andersson S, Slate J (2016) Red carotenoid coloration in the zebra finch is controlled by a cytochrome P450 gene cluster. Curr Biol 26:1435–1440
Nie HT, Jiang KY, Li N, Jahan KF, Jiang LW, Huo ZM, Yan XW (2020) Transcriptome analysis reveals the pigmentation-related genes in two shell color strains of the Manila clam Ruditapes philippinarum. Anim Biotechnol 1–12
Orteu A, Jiggins CD (2020) The genomics of coloration provides insights into adaptive evolution. Nat Rev Genet 21:461–475
Ørom U, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang QH, Guigo R, Shiekhattar R (2010) Long noncoding RNAs with enhancer-like function in human cells. Cell 143:46–58
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL (2015) Stringtie enables improved reconstruction of a transcriptome from rNA-seq reads. Nat Biotechnol 33:290–298
Saenko S, Schilthuizen M (2021) Evo-devo of shell colour in gastropods and bivalves. Curr Opin Genet Dev 69:1–5
Song JL, Wang CD (2019) Transcriptomic and proteomic analyses of genetic factors influencing adductor muscle coloration in QN Orange scallops. BMC Genom 20:363
Starega-Roslan J, Galka-Marciniak P, Krzyzosiak WJ (2015) Nucleotide sequence of miRNA precursor contributes to cleavage site selection by Dicer. Nucleic Acids Res 43:10939–10951
Sun L, Luo HT, Bu DC, Zhao GG, Yu KT, Zhang CH, Liu YN, Chen RS, Zhao Y (2013) Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res 41:e166
Sun XJ, Liu ZH, Zhou LQ, Wu B, Dong YH, Yang AG (2016) Integration of next generation sequencing and EPR analysis to uncover molecular mechanism underlying shell color variation in scallops. PLoS One 11:e0161876
Tarailo-Graovac M, Chen NS (2009) Using repeatMasker to identify UNIT 4.10 repetitive elements in genomic sequences. Curr Protoc Bioinformatics 25(1):4–10
Tian X, Pang XL, Wang LY, Li MG, Dong CJ, Ma X, Wang L, Song DY, Feng JX, Xu P, Li XJ (2018) Dynamic regulation of mRNA and miRNA associated with the developmental stages of skin pigmentation in Japanese ornamental carp. Gene 666:32–43
Vidigal JA, Ventura A (2015) The biological functions of miRNAs: lessons from in vivo studies. Trends Cell Biol 25:137–147
Wang CD, Liu B, Liu X, Ma B, Zhao YM, Zhao X, Liu FQ, Liu GL (2017) Selection of a new scallop strain, the Bohai Red, from the hybrid between the bay scallop and the Peruvian scallop. Aquaculture 479:250–255
Wang LG, Park HJ, Dasari S, Wang SQ, Kocher JP, Li W (2013) CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res 41:e74
Wang LM, Zhu WB, Dong ZJ, Song FB, Dong JJ, Fu JJ (2018) Comparative microRNA-seq analysis depicts candidate miRNAs involved in skin color differentiation in Red Tilapia. Int J Mol Sci 19:1209
Wheeler BM, Heimberg AM, Moy VN, Sperling EA, Holstein TW, Heber S, Petersonb KJ (2009) The deep evolution of metazoan microRNAs. Evol Dev 11:50–68
Williams ST (2017) Molluscan shell colour. Biol Rev 92:1039–1058
Winkler F, Estevez B, Jollan L, Garrido J (2001) Inheritance of the general shell color in the scallop Argopecten purpuratus (Bivalvia: Pectinidae). J Hered 91:521–525
Xu M, Huang J, Shi Y, Zhang H, He MX (2019) Comparative transcriptomic and proteomic analysis of yellow shell and black shell pearl oysters. Pinctada fucata martensii BMC Genom 20:469
Yano M, Nagai K, Morimoto K, Miyamoto H (2006) Shematrin: a family of glycine-rich structural proteins in the shell of the pearl oyster Pinctada fucata. Comp Biochem Physiol B Biochem Mol Biol 144:254–262
Yin HR, Luo MK, Luo WT, Wang LM, Zhu WB, Fu JJ, Dong ZJ (2020) MiR-196a regulates the skin pigmentation of koi carp (Cyprinus carpio L.) by targeting transcription factor mitfa. Aquac Res 52:229–236
Yu SG, Wang G, Liao J, Tang M, Chen J (2020) Identification of key microRNAs affecting melanogenesis of breast muscle in Muchuan black-boned chickens by RNA sequencing. Br Poult Sci 6:225–231
Yu WC, He C, Cai ZQ, Xu F, Wei L, Chen J, Jiang QY, Wei N, Li Z, Guo W, Wang XT (2017) A preliminary study on the pattern, the physiological bases and the molecular mechanism of the adductor muscle scar pigmentation in Pacific oyster Crassostrea gigas. Front Physiol 8:699
Zhang HK, Tan K, Li SK, Ma HY, Zheng HP (2021) The functional roles of the non-coding RNAs in molluscs. Gene 768:145300
Zheng Z, Huang RL, Tian RR, Jiao Y, Du XD (2016) Pm-miR-133 hosting in one potential lncRNA regulates RhoA expression in pearl oyster Pinctada martensii. Gene 591:484–489
Zheng Z, Xiong XW, Zhang JH, Lv SJ, Jiao Y, Deng YW (2019) The global effects of PmRunt co-located and co-expressed with a lincRNA lncRunt in pearl oyster Pinctada fucata martensii. Fish Shellfish Immunol 91:209–215
Zhou L, Chen JH, Li ZZ, Li XX, Hu XD, Huang Y, Zhao XK, Liang CZ, Wang Y, Sun L, Shi M, Xu XH, Shen F, Chen MS, Han ZJ, Peng ZY, Zhai QN (2010) Integrated profiling of microRNAs and mRNAs: microRNAs located on xq27.3 associate with clear cell renal cell carcinoma. Plos One 5:e15224
Zhu XJ, Chen Y, Zhang Z, Zhao SY, Xie LP, Zhang RQ (2020) A species-specific miRNA participates in biomineralization by targeting CDS regions of Prisilkin-39 and ACCBP in Pinctada fucata. Sci Rep 10:8971
Funding
This work was supported by the grants from National Natural Science Foundation of China (31972789 and 31772843), Weihai City (2018NS01), and Industrial Development Project of Qingdao City (20-3-4-16-nsh).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, Z., Li, Q., Liu, S. et al. Integrated Analysis of Coding Genes and Non-coding RNAs Associated with Shell Color in the Pacific Oyster (Crassostrea gigas). Mar Biotechnol 23, 417–429 (2021). https://doi.org/10.1007/s10126-021-10034-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-021-10034-7