
Abstract
The amazing colors and patterns are fascinating characteristics in all of the aquarium species. However, genetic and breeding molecular investigations of ornamental shrimps are rather limited. Here, we present the first transcriptomic analysis and application of microsatellites based on the chromatophore-encoded genes of Neocaridina denticulata to assist freshwater ornamental shrimp germplasm enhancement and its extensive applications. A total of 65,402 unigenes were annotated, and 4706 differentially expressed genes were screened and identified between super red shrimp and chocolate shrimp strains. Several gene ratios were examined to put in perspective possible genetic markers for the different strains of normal pigmentation development, including flotillin-2-like, keratin, the G protein–coupled receptor Mth2-like, annexin A7, and unconventional myosin-IXb-like. Five simple sequence repeat markers were effective for colored shrimps and were used to develop a marker-assisted selection platform for systematic breeding management program to maintain genetic diversity of the species. These markers could also be used to assist the identification of pure strains and increase the genetic stability of ornamental shrimp color phenotypes. Consequently, our results of microsatellite marker development are valuable for assisting shrimp genetic and selection breeding studies on freshwater ornamental shrimp and related crystal shrimp species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ornamental aquatic species have ever-changing and attractive external appearances, shapes, and vivid colors (Ertl et al. 2013). The number of artificially cultivated varieties continues to expand, and innovations in breeding techniques and equipment and popularity of feed nutrition science as well as convenience of transportation have been the primary factors promoting the rapid worldwide development of the ornamental aquatic animal industry (Nguyen et al. 2014; Negisho et al. 2019; Pinnegar and Murray 2019).
Freshwater shrimp Neocaridina denticulata is a decapod crustacean ornamental species originated in Taiwan. Initially, traditional selective breeding methods were used by the industry to produce shrimp with high ornamental and economic value through long-term genetic improvements from black shell shrimp, which was used as fish fodder and sold by kilogram (Hung et al. 1993; Ariyanathan and Serebiah 2016). Owing to their small size, ease of raising, short life cycle, convenient to transport, and high productivity, these shrimp have been welcomed by many aquatic animal lovers in developed countries (Nur and Christianus 2013; Mykles and Hui 2015). In addition to adding esthetic value to aquarium landscapes, shrimp can consume residual food and clear algae from water, thereby promoting ecological stability and becoming a popular item in small aquaria worldwide (Patoka et al. 2015; Levitt-Barmats et al. 2019).
From breeding experience and long-term observation of commercially available colored shrimp, subtle genetic variations in multiple ornamental traits have been developed by breeding different strains (Levitt-Barmats et al. 2019). Currently, colored shrimp can be divided into red, orange, yellow, green, blue, indigo, black, and white strains, including super red, red rili, blue velvet, chocolate, sunkist, snowball, snow white, and over 20 colored commercial strains, with continuous addition of more strains.
Understanding the mechanisms responsible for the development of color and patterning in ornamental aquatic animals can effectively enable breeders to increase the heritability of color traits for genetic breeding, achieve a more stable production, or develop more varied novel commercial strains, and has significance for their physiology and behavior (McNamara and Milograna 2015). In recent years, color polymorphism has also received attention for its impact on speciation and adaptation (Yue et al. 2015). Color changes are primarily regulated by substances secreted from radially branching chromatophores located in epidermal connective tissue and differentiated from neural crest cells (Wade et al. 2008). A variety of chromatophores contain granules of white, red, yellow, blue, brown, and black pigments, which originate from carotenoids in food. These pigments bind to proteins, and chromatophores exhibit different degrees of dispersion and accumulation, and thus produce different colors. However, no scientific research has yet been reported on the molecular regulation mechanisms or genetic markers of body surface color formation of freshwater ornamental shrimp.
In a study of cold-blooded animals, five types of chromatophores, i.e., melanophores, erythrophores, xanthophores, iridophores, and leucophores, were determined to contain different pigments and have different functions (Ben et al. 2003). Among them, melanophores contain melanin, which primarily manifests as black or brown body color; xanthophores and erythrophores primarily contain pteridine and carotenoid and manifest as red, orange, or yellow body color, while iridophores and leucophores contain guanine and other purines and produce lustrous metallic colors such as blue, green, and white through light reflection (Lynn Lamoreux et al. 2005).
Body pigment development is mainly divided into two stages (Woolley et al. 2014): during embryonic development, neural crest cells migrate and differentiate into different types of melanophores, xanthophores, and iridophores under specific regulation of the genes Kit (kit type III receptor tyrosine kinase) (Parichy et al. 1999; Kottler et al. 2013), Ednrb (endothelin receptor B) (Parichy et al. 2000), Mitf (microphthalmia-associated transcription factor), and pnp4a (purine nucleoside phosphorylase) (Curran et al. 2010) (1), and chromatophores produce specific pigments through metabolic processes (2).
With the rapid development of molecular biology and genomics research methods, next-generation sequencing (NGS) platforms can generate genomic DNA sequence libraries of various species of ornamental aquatic organisms (Ghaffari et al. 2014; Kenny et al. 2014; Xu et al. 2014; Yu et al. 2014; Sin et al. 2015). Moreover, DNA molecular markers have been used to identify new strains of ornamental aquatic organisms and to select color-related properties (Yue and Chang 2010). From the analysis of genetic variation between the transcriptomes of the Fenneropenaeus merguiensis and the Macrobrachium olfersi by NGS technology and microarray analysis, the Ca2+/cGMP signaling pathways were found to participate in pigment aggregation, with the carotenoid astaxanthin, crustacyanin, red pigment concentrating hormone (RPCH), and G protein–coupled receptor (GPCR) factors being differentially expressed (Ertl et al. 2013; Milograna et al. 2014, 2016).
The objective of this study was to reveal differential expression genes from the transcriptome database between two pure strains of the freshwater ornamental shrimps N. denticulata, i.e., light-colored super red shrimp (SRS) and dark-colored chocolate shrimp (CS), by using the (NGS) platform, and to explore biological pathways and develop molecular markers involving the putative chromatophore-encoded functional genes. Furthermore, these analyses may not only provide new insight into the causes of coloration in colored ornamental shrimp but also enable breeders to pursue higher quality, more stable heritability of chromogenic phenotypic traits, and even create novel attractive varieties for trade in the future.
Materials and Methods
Experimental Shrimp, Genomic DNA Extraction
Experimental colored shrimp samples were obtained from Larmax International Co., Ltd. in southern Taiwan, including SRS and CS. The native strain black shell shrimp (BSS) was used as the experimental control.
Each shrimp strain was separately reared in a smart breeding cycle system containing a 3- or 10-L feeding cylinder with independent drainage, aeration, and a water flow of approximately 0.12 L/h. Water quality was maintained by a filter circulation system with temperature, pH, and general hardness (GH) controlled at 22–25 °C, 6.5–7.0, and 3.0–4.0, respectively.
The MasterPure™ DNA Purification Kit (Epicenter, Madison, USA) was used to extract genomic DNA from shrimp. Genomic DNA was diluted to 25–40 ng/μL and samples were labeled stating the tissue number to be tested, sample name, extraction date, and other information related to the origin of the sample. Samples were stored at − 20 °C for use in subsequent PCR analyses.
Total RNA Extraction
The experimental animals were 1–4-week-old SRS and CS. Total RNA was extracted using EasyPure Total RNA Spin Kit (Bioman, Taipei, Taiwan).
Fresh shrimp tissue samples were collected and placed in a 1.5-mL microcentrifuge tube containing 500 μL RNA TriPure Isolation Reagent (Roche Applied Science, Germany). Three stainless steel beads (3 mm) and a 1–5-mm steel bead (LabTurbo®) were added to the tube and the mixture was placed in a SpeedMill PLUS high-speed tissue homogenizer (Analytik Jena AG). The sample was disrupted three times for 1 min each and placed at room temperature for 5 min. The tissue homogenate was placed in a 2-mL filter column and centrifuged at 10,000×g for 2 min at 4 °C. The filtrate was collected and placed in a new 1.5-mL microcentrifuge tube, and 400 μL 70% ethanol was added and mixed. After discarding the liquid in the collection tube, the RB column was placed in a 1.5-mL microcentrifuge tube. RNase-free water (50 μL) was placed in the RB column, which was allowed to stand for 5 min until the RNAse-free water was completely absorbed, and centrifuged at 10,000×g for 2 min at 4 °C. Purified total RNA was collected and the MaestroNano spectrophotometer (Maestrogen, Las Vegas, NV, USA) was used to measure optical density (OD260 and OD280) and calculate the concentration.
High-Throughput Next-Generation Transcriptome Sequencing
Samples of total RNA (10 μg) from SRS and CS strains were used to determine RNA quality and to construct a transcriptome gene library. mRNA was enriched by using the oligo (dT) magnetic beads. Mixed with the fragmentation buffer, mRNA was divided into short fragments. Then, the first strand of cDNA was synthesized by using random hexamer-primer, and buffer, dNTPs, RNase H, and DNA polymerase I were added to synthesize the second strand. The double strand cDNA was purified with magnetic beads. Then, 3′-end single nucleotide A (adenine) addition was performed. Finally, sequencing adaptors were ligated to the fragments and these were enriched by PCR amplification. During the quality control step, Agilent 2100 Bioanaylzer (Agilent Technologies, USA) and ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, USA) were used to qualify and quantify the sample library.
The Illumina HiSeq 2000 NGS platform was used to analyze sequences and data on differential gene expression (Lee 2017). The premise of NGS was the fragmentation of total RNA followed by reverse transcription into cDNA, along with the addition of adaptors and primers before sequencing. The transcriptome de novo assembly of high-quality sequence read data was achieved with Trinity (Fu et al. 2012) short read assembly software (http://trinityrnaseq.sourceforge.net) and with the Japanese swamp shrimp native species (N. denticulata) provided by the BioProject database on NCBI (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA240382), screening of unigenes with NCBI non-redundant protein (Nr), Gene Ontology (GO) (E-value < 10−5) (Conesa et al. 2005), Clusters of Orthologous Groups (COG), and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al. 2008) pathway enrichment analysis. Blast2GO (v2.5.0) was used to obtain GO annotations (E-value < 10−5) on the basis of the Nr notes. In addition, unigenes were functionally aligned and annotated using GO/KEGG/COG annotation, and coding proteins were predicted and classified according to the COG database. Molecular marker gene locus detection online software (www.genome.jp/kegg/kegg4.html) was used to search for appropriate simple sequence repeat (SSR) markers to screen and search against the KOG database through BLASTX and KGGG pathways for color properties in ornamental shrimp.
An NGS platform was used to compare with unigene databases and annotations, including unigene protein and COG functional annotations. First, unigene sequences were compared using BLASTX; then, the NR gene (Fig. S1) and Swiss-Prot protein databases were used. The Swiss-Prot database is a database of screened proteins, KEGG is a database used to analyze the metabolic pathways and functions of gene products, and COG is a database used to directly compare phylogeny (E-value < 10−5) (Fig. S2). The two databases were compared; unigenes were compared with the Nt nucleotide database using BLASTN (E-value < 10−5), and the proteins with the highest similarity to unigenes were obtained, thereby providing protein functional annotation information on those unigenes. Next, these unigenes were compared with the NR, NT, Swiss-Prot, KEGG, COG, and GO databases (Fig. S3), and the unigenes were subjected to further annotation.
Differential Analysis of Gene Expression
To establish the maximum and minimum values of the log2 ratio (CS/SRS) between the expression of transcriptome library genes in the SRS and CS strains of N. denticulata, differentially expressed genes (DEGs) were investigated between different samples, as shown by fragments per kilobase of exon per million fragments mapped (FPKM).
FPKM is calculated from the following equation:
where C is the number of uniquely matched gene fragments, N is the total number of uniquely matched gene fragments, and L is the number of bases in the gene (Mortazavi et al. 2008).
Analysis of Microsatellite Markers
MicroSAtellite (MISA) (Thiel et al. 2003) molecular marker gene locus prediction software was used to detect DEGs and identify microsatellite markers. Microsatellite markers of at least 150 bp were screened; ClustVis online software was used for pattern clustering analysis of large amounts of data obtained from different strains. The data were presented as a heat map. Twenty-nine microsatellite markers were identified in this manner and used for subsequent analyses.
Twenty-six microsatellite markers were used to screen color properties in four offspring groups of P1 [SRS (female) × SRS (male)], P2 [CS (female) × CS (male)], H1 [SRS (female) × CS (male)], and H2 [CS (female) × SRS (male)] on 50 shrimps from each group (Fig. S4). Multiple fluorescence labelling was used during the first PCR amplification. A forward primer containing an adaptor was annealed to gDNA fragments. PCR was performed using a 96-well Veriti® thermal cycler (Applied Biosystems Life Technologiess, Carlsbad, CA, USA). The eight microsatellite markers were amplified a second time using fluorescent forward primers. PCR products of all samples were separated using 2% agarose gel electrophoresis. To reduce the probability of dual-allele gene recognition errors, PCR was performed in duplicate for each sample, and the microsatellite genotype of the sample was confirmed if both duplicates obtained the same results. The four PCR products with different fluorescent labels were mixed thoroughly. The capillary electrophoresis instrument ABI PRISM® 3730xl automated DNA Analyzer (Applied Biosystems, USA) was used to separate the SSR fragments of each sample. The output was analyzed using GeneMapper software (versions 4.0, Applied Biosystems).
Statistical Analysis
Geneious software (v6.1.6) package (http://www.geneious.com/) was used to interpret and analyze multiple fluorescent PCR polymorphic marker genotypes of all microsatellite marker data. Then, multi-allelic data scorings (A, B, C, etc.) were imported into POPGENE32 software version 1.32 (Yeh et al. 2000) for the statistical analysis of parameters, including the number of alleles (Na), allele frequency (Ne), and population diversity measures, i.e., observed heterozygosity (Ho), expected heterozygosity (He), polymorphism information content (PIC), and fixation index (FIS), of each microsatellite gene locus (Pan and Yang 2010). Population diversity measures were calculated as:
where Nhet is the number of heterozygous individuals and Nhom is the number of homozygous individuals.
where n is the number of allele at each locus, and Pi is the frequency of the ith gene (Nei 1978).
where n is the number of alleles and Pi and Pj are the frequencies of the ith and jth alleles, respectively (Botstein et al. 1980).
IBM SPSS Statistics v22.0.0 software was used to determine whether the genotypes were significantly correlated with the p values obtained from strain analysis, and M-ANOVA was used to test multiple variables.
Results
High-Throughput Next-Generation Transcriptome Sequencing
For high-throughput NGS of ornamental shrimp transcriptome, three 1–4-week-old SRS and three 1–4-week-old CS were obtained from Larmax International Co., Ltd. for transcriptomic analysis. Using the Illumina HiSeq 2000 sequencing platform, a total of 9,838,273,000 nt of data were obtained from the SRS and CS strains (Table 1). After assembly of transcriptome contigs, 105,892 unigenes with a total length of 125,528,321 nt were obtained, with an average length and N50 of 1185 and 3254 nt, respectively (Table 1). Overall, 36,665, 18,150, 30,864, 27,751, 15,407, and 12,642 unigenes were annotated in NR, NT, Swiss-Prot, KEGG, COG, and GO, respectively, and a total of 65,402 unigenes were annotated common to all strains (Table 2) (Table S1-5).
Analysis of Different Gene Expression in Colored Shrimp
A Venn diagram of genes commonly expressed found in colored shrimp and of specific genes (Fig. 1) showed that SRS, CS, and JSS have 65,402 genes in common, with 2317 and 2228 genes specific to SRS and CS, respectively. All unigenes were classified as high-expressed, low-expressed, or similar (Fig. 2a). Using the log2 ratio (CS/SRS) between expression levels in CS and SRS, 2230 and 2476 genes were classed as high- and low-expressed, respectively, in SRS relative to CS (Fig. 2b). KEGG pathway enrichment analysis of the 20 distinct metabolic pathways identified based on DEGs between SRS and CS is listed in Fig. 3. Among them, the top five pathways were amoebiasis (ko05146), vibrio cholera infection (ko05110), salivary secretion (ko04970), protein digestion and absorption (ko04974), and dorso-ventral axis formation (ko04320) (Fig. 3). Furthermore, a significant proportion of sequences in our transcriptome were involved in the function classification of “general function prediction only” based on both COG and KEGG databases, and the “replication, recombination, and repair,” “translation, ribosomal structure and biogenesis,” “transcription,” “carbohydrate transport and metabolism,” and “signal transduction mechanisms” pathways were detected (Fig. S2). Among DEGs (Fig. 4), polymorphic gene–based SSR markers were screened and used for subsequent genetic diversity analyses (Table S6).

Venn diagram showing relationships between the three transcriptome datasets, i.e., Japanese swamp shrimp (JSS), super red shrimp (SRS), and chocolate shrimp (CS). Numbers in parentheses represent the total number of expressed genes in each strain. A total of 65,402 unigenes were annotated common to all strains

Comparison of unigene expression level between super red shrimp (SRS) and chocolate shrimp (CS) of N. denticulata. a Scatter plot analysis of significant differential expression level between SRS and CS. X-axis, gene expression of SRS; Y-axis, gene expression of CS. FDR < 0.001 and the absolute value of log2 fold change ≥ 1 were used as the threshold to judge the significance of gene expression differences. Blue and yellow dots indicate the differentially expressed unigenes, and brown dots indicate unigenes that were not differentially expressed between SRS and CS. b The numbers of differentially expressed genes high- (yellow) and low-regulated (blue) in SRS versus CS

Scatterplot for Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway enrichment analysis of identified differentially expressed genes (DEGs) between SRS and CS strains. Rich factor represents the ratio of differentially expressed gene numbers annotated in this pathway term to all gene numbers annotated in this pathway term. Greater rich factor means greater intensiveness. Q-value is the corrected p value ranging from 0 to 1, with lower values representing greater intensiveness. The top 20 pathway entries in the degree of enrichment were shown. The differentially expressed genes between SRS and CS strains were identified to be involved in 20 distinct metabolic pathways. Among them, the top five pathways were amoebiasis (ko05146), vibrio cholera infection (ko05110), salivary secretion (ko04970), protein digestion and absorption (ko04974), and dorso-ventral axis formation (ko04320)

Heat map representing functional gene expression of N. denticulata transcripts containing microsatellite locus in super red shrimp (SRS) and chocolate shrimp (CS) based on their relative fragments per kilobase of exon per million fragments mapped (FPKM) values. Transcripts were hierarchically cluster based on correlation distance and average linkage method. Red and green indicate high and low level of expression, respectively. FPKM, fragments per kilobase of transcript per million mapped reads
Microsatellite Software Analysis
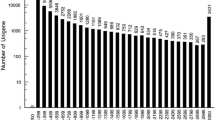
Using MISA analysis software, 25,355 microsatellite markers containing one, two, three, four, five, or six base repeats (Table 3, Fig. 5) were found. Overall, 7051 (27.81%), 7589 (29.93%), 9076 (35.8%), 1352 (5.33%), 167 (0.66%), and 120 (0.47%) microsatellite markers had repeats comprising one, two, three, four, five, and six bases, respectively. The most common microsatellites with repeats of three bases were AAT/ATT, AAG/CTT, AAG/CCT, and ATC/GAT with 2394 (26.38%), 1299 (14.31%), 1243 (13.70%), and 1174 (12.94%), respectively. The most common microsatellites with repeats of two bases were AC/GT, AG/CT, and AT/TA, with 2283 (30.08%), 3160 (41.64%), and 2131 (28.08%), respectively.

The percentage distribution of the total number of 18,304 perfect microsatellite SSRs among different nucleotide classes of dimers, trimers, quadmers, pentamers, and hexamers motif sequences, repeated nucleotide types, and repeat number found in the transcriptome of N. denticulata
By screening DEGs and using bioinformatics analyses, including GO annotations, pathway enrichment analysis, gene locus prediction software, functional clustering, differential expression, and microsatellite (SSR) marker mutant sequencing, 26 possible molecular markers of color-related functional genes were found in colored shrimp, i.e., CL1904_4, CL2407_4, CL2408_7, CL2911_2, CL32_6, CL3384_1, CL3484_2, CL3550_2, CL3563_2, CL3771_4, CL4393_2, CL5162_2, CL5500_1, CL5558_3, CL621_12, CL6971_3, CL85_6, Unigene10624, Unigene14740, Unigene19134, Unigene20167, Unigene28786, Unigene31684, Unigene32861, Unigene36045, and Unigene64263. ClustVis was used for pattern clustering analysis of DEGs, and heat map analysis of these genes in both shrimp strains (Fig. 4) revealed that the 26 markers were differentially expressed between the two strains.
SRS and CS Microsatellite Marker Analysis
Transcriptome sequencing platform, MISA software analysis, and heat map analysis, identified 26 microsatellite markers that were correlated with DEGs (Table S7). Among these, 16 feasible markers were identified, from which (Table 4), the five functional genes correlated with color properties of colored shrimp, i.e., Unigene32861, Unigene28786, CL5162_2, Unigene14740, and Unigene10624, were screened, corresponding to flotillin-2-like, keratin (type I cytoskeletal 19), GPCR Mth2-like, annexin A7, and unconventional myosin-IXb-like, respectively. Short tandem repeat fragments of multiple fluorescent markers of alleles were arranged by capillary electrophoresis into A, B, and C ordered by increasing size. The frequency of each allele was calculated and POPGENE32 software was used to analyze population genetic diversity. Because all markers were type I functional markers and the number of alleles was small, the Unigene14740 genotype was identified as the most common among all markers, whereas the Unigene10624 genotype was the least common. In SRS shrimp, the average observed and expected heterozygosity among these five markers was 0.40 ± 0.45 and 0.35 ± 0.32, respectively, while the average PIC and FIS was 0.34 ± 0.32 and 0.14 ± 0.27, respectively (Table 5). In CS shrimp, the average observed and expected heterozygosity among these five markers was 0.47 ± 0.3 and 0.45 ± 0.24, respectively, while the average PIC and FIS was 0.44 ± 0.24 and − 0.09 ± 0.33, respectively (Table 5).
Analysis of Correlations Between Marker Genes and Strain
Regarding the flotillin-2-like gene (Unigene32861 locus), the AA genotype of the 3′-UTR in the CS group was significantly higher than that in the SRS group (p < 0.05). In the SRS strain, the alleles were C > B > A, whereas in the CS strain, the alleles were A > C > B. The genotype frequencies of AC and BC in the SRS strain were 0.34 and 0.53, respectively, and the genotype frequency of AA in the CS strain was 0.81. Moreover, the genotypes were significantly correlated with strain (p < 0.01).
Regarding the keratin (type I cytoskeletal 19) (Unigene28786 locus), the frequency of CC genotype in the SRS group was significantly higher than that in the CS group (p < 0.05). In the SRS strain, the alleles are C > B = A, whereas in the CS strain, the allele frequency were C > B > A, making the CC genotype frequency in the SRS strain 0.9, and those of AC, BC, and CC in the CS strain 0.25, 0.28, and 0.41, respectively; the genotypes were significantly correlated with strain (p < 0.01).
Regarding the GPCR Mth2-like (CL5162 locus), the AA genotype frequency in the SRS group was significantly higher than that in the CS group (p < 0.05). Although the allele frequencies in SRS and CS strains were the same (A > B > C), the genotype frequencies of AA in the SRS strain was 0.78, and those of AA and AC in the CS strain were 0.34 and 0.22, respectively; the genotypes were significantly correlated with strain (p < 0.01). Regarding the annexin A7 (Unigene14740 locus), the BC genotype frequency was significantly in the CS group higher than that in the SRS group (p < 0.05). In the SRS strain, the alleles were D > A > C > B > E, whereas in the CS strain, the alleles were C > B = D > A = E; CS does not carry A or E alleles. Thus, the genotype frequencies of CD and AD in the SRS strain were 0.38 and 0.31, respectively, and those of BC and CD in the CS strain 0.5 and 0.31, respectively. The AA, AD, AE, and BC genotypes were significantly correlated with strain (p < 0.01), and the BB and DE genotypes were significantly correlated with strain (p < 0.05).
Regarding the myosin-IXb-like (Unigene10624 locus), the AA genotype frequency in the SRS group was significantly higher than that in the CS group (p < 0.05). In the SRS and CS strain, the alleles were A > B, with genotype frequencies of AA in the SRS strain of 1.00, while those of AA and AB in the CS strain were 0.78 and 0.22, respectively. AA and AB genotypes were significantly correlated with strain (p < 0.01).
Marker analysis of the SRS and CS strains revealed that SRS-specific genetic markers, including the BC genotype of flotillin-2-like and the AA, AD, AE, BB, and DE genotypes of annexin A7, were significantly correlated with the SRS strain (p < 0.01). CS-specific genetic markers, including the BC genotype of keratin, the BC, CC, and AC genotypes of GPCR Mth2-like, the BC genotype of annexin A7, and the AB genotype of myosin-IXb-like, were significantly correlated with the CS strain (p < 0.01) (Table 6).
Discussion
N. denticulata is an ornamental shrimp with a high added-value product (Patoka et al. 2015; Levitt-Barmats et al. 2019). Studies on N. denticulata have mainly focused on molecular phylogeny (Shih and Cai 2007; von Rintelen et al. 2012), environmental adaptations (Suzuki and Kanou 2014), microbiota diversity (Cheung et al. 2015; Cornejo-Granados et al. 2018), and environmental toxicological and pharmacological applications (Sung et al. 2014; Wu and Li 2015; Hu et al. 2019). However, the studies focusing on molecular marker development and genetic diversity are limited, and there is still no transcriptome analysis report about this species, which blocks the in-depth study on N. denticulata (Mykles and Hui 2015; Nong et al. 2020).
RNA-Seq is an appropriate tool for obtaining transcriptome data widely applied in various organisms (Mortazavi et al. 2008; Leu et al. 2011; Ghaffari et al. 2014; Chen et al. 2015; Yue et al. 2015; Wang et al. 2018). This study presents the first genetic and breeding research on whole organ tissues of N. denticulata through transcriptome sequencing. Notably, a large number of N. denticulata non-redundant unigenes (105,892) were generated with the Illumina HiSeq 2000 platform, and numerous expressed sequence tags (ESTs) were available. Among the identified unigenes, 65,402 (61.76%) were successfully annotated through BLAST searching against the public Nr, GO, COG, KOG, and KEGG databases. GO and COG analyses revealed the distribution of functional genes in N. denticulata, while KEGG database searching successfully revealed the functions of cellular-process genes and the gene products of metabolic processes. With a high-quality N. denticulata transcriptome assembly, the unigenes developed in this study would enable genetic studies of these freshwater ornamental shrimps, which would enrich our understanding of their regulating mechanism for color-related genes and genetic diversity. Additionally, this is crucial for the management planning of genetic improvements in this ornamental species.
The functional classification of these transcripts according to the GO database showed that “binding” and “catalytic activity” were the dominant molecular functions (Fig. S3), which was consistent with previous studies in other crustaceans (Leu et al. 2011; Ertl et al. 2013; Wang et al. 2019; Tang et al. 2020). This reflected that actin and tubulin secreted by epithelial cells to the extracellular matrix and a signaling molecule activated a specific receptor located on the cell surface or inside the cell, and triggered a biochemical chain of events in the accumulation and dispersion of pigment granules among the chromatophores (Ertl et al. 2013). Therefore, it is hypothesized that there are many complex interactions between different pigment cells of chromatophores, which is potentially involved in the formation of variant body coloration of N. denticulata.
Previous studies have identified various gene products in crustaceans that regulate coloration, including crustacyanin, sarcoplasmic calcium-binding protein, forms of actin, RPCH, cyclic AMP (cAMP), pigment dispersing hormone (α-PDH), and cAMP-dependent protein kinase (Ertl et al. 2013; Milograna et al. 2016). Our transcript analysis of these gene products is consistent with the unigene-enrichment in “secondary metabolite biosynthesis, transport and catabolism.”
Molecular marker technology can be used to detect and reflect genetic differences at the genomic DNA level with individual specificity. It has the advantage of environmental stability (Chow et al. 2018) and is commonly used as an important molecular tool for assessing genetic diversity (Sajeela et al. 2019; Zhao et al. 2019). Among several kinds of molecular markers, SSR techniques have the advantages of high polymorphism, superior repeatability, and widespread application. Moreover, it has been widely applied in the field of aquatic breeding and genetic management in recent years (Andriantahina et al. 2013). For the genetic improvement of aquaculture species, research on shrimp germplasm resources has developed and progressed from analysis of phenotype to genotype (Nong et al. 2020).
RNA-sequencing is considered an effective way to acquire EST sequences for identifying novel genes and developing SSR markers (Chen et al. 2015; Wang et al. 2018). In this study, the overall analysis strategy was mainly to obtain information about the regulation of biological functions of genes and color-related SSR markers from the whole organ tissues of different and monotonously colored-shell ornamental shrimp strains, i.e., SRS (light-colored) and CS (dark-colored), by using RNA-Seq technology. A comparative transcriptomic analysis was performed to reveal the transcriptional alterations in SRS and CS strains.
All 65,402 unigenes were used to detect SSRs, and a total of 25,355 (38.77%) SSRs were identified, which is a lower percentage than those in banana shrimp (Fenneropenaeus merguiensis) (43.5%) (Wang et al. 2017), and giant freshwater prawns (Macrobrachium rosenbergii) (48.76%) (Jiang et al. 2019), but higher than that in ridgetail white prawn (Exopalaemon carinicauda) (10.97%) (Wang et al. 2018). On average, the SSR loci were found at a distance of 4.95 kbp, which is higher than those in F. merguiensis (1.02 kbp), and M. rosenbergii (0.93 kbp), but lower than that in E. carinicauda (6.6 kbp). Among the mined SSRs, the proportion of mono- (7051, 27.81%), di- (7589, 29.93%) and tri- (9076, 35.8%) nucleotide accounted for more than 93%. This result was consistent with those for other crustacean species (Wang et al. 2017; Wang et al. 2018; Jiang et al. 2019).
According to multiple metabolic pathways with biological functions, such as amoebiasis (ko05146), vibrio cholera infection (ko05110), salivary secretion (ko04970), protein digestion and absorption (ko04974), dorso-ventral axis formation (ko04320), insulin signaling pathway (ko04910), and regulation of actin cytoskeleton (ko04810), from the transcriptome databank, we identified EST-SSR markers in candidate genes, such as flotilin-2-like (Neumann-Giesen et al. 2007), keratin or type I cytoskeletal 19 (Ertl et al. 2013), GPCR Mth2-like (Ha et al. 2003), annexin A7 (Castle et al. 2003), and unconventional myosin (myosin-IXb-like) (Wirth et al. 1996; Chieregatti et al. 1998). These genes have been shown to be involved in phosphorylation of multiple tyrosines and endocytosis (Riento et al. 2009), pigment particle transport (Ha et al. 2003), melanin content and type in melanocytes (Tuma and Gelfand 1999; Van der Salm et al. 2005; Ertl et al. 2013), cAMP signal transduction pathway activated by GPCR (García-Borrón et al. 2005), calcium-dependent phospholipids binding proteins (Brownawell and Creutz 1997), membrane-associated protein distributions that are involved in a variety of cell functions (McMichael et al. 2014), production of specific pigments, or chromatophore formation and transformation through metabolic processes. Based on a mixed linear model, an association analysis of hybridization experiment was performed on 200 F1 progeny individuals from two groups of pure strain (SRS × SRS, CS × CS) and two reciprocal hybrid strains (SRS × CS, CS × SRS) of SRS and CS. The genetic diversity level of SRS and CS N. denticulata pure and hybrid germplasm resources was validated with fluorescently labeled capillary electrophoresis.
Previous studies have explored EST-SSR polymorphism results in Epinephelus lanceolatus (Zeng et al. 2008), Megalobrama pellegrini (Wang et al. 2012), and Pelteobagrus fulvidraco (Zhang et al. 2014), finding that the polymorphism level is low, medium, and high when PIC is 0–0.25, 0.25–0.5, and > 0.5, respectively (Zeng et al. 2008). However, the SSR markers of five functional genes in N. denticulata produced 16 alleles (numbered between 2 and 5) and 25 genotypes, with an average of 3.8 alleles per locus (Na) and PIC of 0.34–0.44, showing a medium polymorphism level. These results may be attributed to the expected number of SSRs and polymorphisms in DNA protein-coding sequences being lower than that of non-coding sequences, and the mutation rate within these regions being lower than that in other DNA sequences. According to the theory, one polymorphic locus in this study was a high-level polymorphic locus, and four of them were medium-level polymorphic loci. These results indicated that 5 out of 26 primer pairs in this study should be used for genetic diversity analysis of N. denticulata. Genetic diversity analysis showed that most N. denticulata resources clustered according to origin area, indicating that geographic variation is a significant cause of germplasm variation and genetic diversity. The false positive rate of color phenotype identification (misrecognition) of the single and combined markers in the two strains was lower than 5% (4.31%) and 0.01% (0.0018%), respectively.
The genetic polymorphism coefficient and correlation analysis were used to distinguish the four breeding resources of SRS and CS (Chen et al. 2017). The light-colored SRS presented a higher percentage of homozygous genotypes in genes such as keratin, GPCR Mth2-like, and myosin-IXb-like than that in CS. Meanwhile, the dark-colored CS presented a higher percentage of homozygous genotypes in flotillin-2-like genes than that in light-colored SRS, which were dominated by homozygotes. This phenomenon can explain why CS has more potential for selection and purifying efficiency than SRS in the genetic improvement of color (Chen et al. 2017).
Coloration is affected and regulated by multiple genes and environmental factors. Ornamental shrimp transcriptome libraries are important for studying the regulatory basis of color-related properties in colored or crystal shrimp. These findings will represent a breakthrough for the confirmation of freshwater ornamental shrimp strains, and will benefit research on color-related gene expression and on the basic mechanisms regulating pigment deposition (Yamada et al. 1990; Wade et al. 2012). In addition, they provide genetic comparison and reference value data for other related invertebrates. Therefore, in addition to testing different heterozygous strains and offspring to identify multiple microsatellite markers related to color properties, future identification of genetic traits can be performed using a set of appropriate DNA barcodes by comparing whole genome sequences.
A combination of specific genotypes can serve as indicators for the improvement of molecular marker-assisted screening, allowing the establishment of standard operation models for molecular selective breeding techniques. Results generated through research and development have potential application and economic value in the development and modernization of the ornamental shrimp industry, and provide industries with more complete and scientific means to produce, market, and certify brands, thus introducing a new element to the ornamental shrimp aquaculture industry. In the future, this is expected to continue to enhance the ornamental aquarium and biotechnology industry’s international market to create new opportunities.
References
Andriantahina F, Liu X, Huang H (2013) Genetic map construction and quantitative trait locus (QTL) detection of growth-related traits in Litopenaeus vannamei for selective breeding applications. PLoS One 8:e75206
Ariyanathan G, Serebiah JS (2016) Selective and interbreeding of freshwater ornamental shrimps with endemic aquarium plants. Int J Curr Res Biosci Plant Biol 3:144–150
Ben J, Lim TM, Phang VP, Chan WK (2003) Cloning and tissue expression of 6-pyruvoyl tetrahydropterin synthase and xanthine dehydrogenase from Poecilia reticulata. Mar Biotechnol 5:568–578
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Brownawell AM, Creutz CE (1997) Calcium-dependent binding of sorcin to the N-terminal domain of synexin (annexin VII). J Biol Chem 272:22182–22190
Castle J, Garrett-Engele P, Armour CD, Duenwald SJ, Loerch PM, Meyer MR, Schadt EE, Stoughton R, Parrish ML, Shoemaker DD, Johnson JM (2003) Optimization of oligonucleotide arrays and RNA amplification protocols for analysis of transcript structure and alternative splicing. Genome Biol 4:1
Chen X, Mei J, Wu J, Jing J, Ma W, Zhang J, Dan C, Wang W, Gui JF (2015) A comprehensive transcriptome provides candidate genes for sex determination/differentiation and SSR/SNP markers in yellow catfish. Mar Biotechnol 17:190–198
Chen J, Glémin S, Lascoux M (2017) Genetic diversity and the efficacy of purifying selection across plant and animal species. Mol Biol Evol 34:1417–1428
Cheung MK, Yip HY, Nong W, Law PT, Chu KH, Kwan HS, Hui JH (2015) Rapid change of microbiota diversity in the gut but not the hepatopancreas during gonadal development of the new shrimp model Neocaridina denticulata. Mar Biotechnol 17:811–819
Chieregatti E, Gärtner A, Stöffler HE, Bähler M (1998) Myr 7 is a novel myosin IX-RhoGAP expressed in rat brain. J Cell Sci 111:3597–3608
Chow LH, Ma KY, Hui JHL, Chu KH (2018) Isolation and characterization of polymorphic microsatellite loci for Caridina cantonensis and transferability across eight confamilial species (Atyidae, Decapoda). Zool Stud 57:e19
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Cornejo-Granados F, Gallardo-Becerra L, Leonardo-Reza M, Ochoa-Romo JP, Ochoa-Leyva A (2018) A meta-analysis reveals the environmental and host factors shaping the structure and function of the shrimp microbiota. PeerJ 6:e5382
Curran K, Lister JA, Kunkel GR, Prendergast A, Parichy DM, Raible DW (2010) Interplay between Foxd3 and Mitf regulates cell fate plasticity in the zebrafish neural crest. Dev Biol 344:107–118
Ertl NG, Elizur A, Brooks P, Kuballa AV, Anderson TA, Knibb WR (2013) Molecular characterisation of colour formation in the prawn Fenneropenaeus merguiensis. PLoS One 8:e56920
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150-3152
García-Borrón JC, Sánchez-Laorden BL, Jiménez-Cervantes C (2005) Melanocortin-1 receptor structure and functional regulation. Pigment Cell Res 18:393–410
Ghaffari N, Sanchez-Flores A, Doan R, Garcia-Orozco KD, Chen PL, Ochoa-Leyva A, Lopez-Zavala AA, Carrasco JS, Hong C, Brieba LG, Rudiño-Piñera E, Blood PD, Sawyer JE, Johnson CD, Dindot SV, Sotelo-Mundo RR, Criscitiello MF (2014) Novel transcriptome assembly and improved annotation of the whiteleg shrimp (Litopenaeus vannamei), a dominant crustacean in global seafood mariculture. Sci Rep 4:7081
Ha T, Naysmith L, Waterston K, Oh C, Weller R, Rees JL (2003) Defining the quantitative contribution of the melanocortin 1 receptor (MC1R) to variation in pigmentary phenotype. Ann N Y Acad Sci 994:339–347
Hu XL, Niu JJ, Meng Q, Chai YH, Chu KH, Chan KM (2019) Effects of two juvenile hormone analogue insecticides, fenoxycarb and methoprene, on Neocaridina davidi. Environ Pollut 253:89–99
Hung MS, Chan TY, Yu HP (1993) Atyid shrimps (Decapoda: Caridea) of Taiwan, with descriptions of three new species. J Crustac Biol 13:481–503
Jiang J, Yuan X, Qiu Q, Huang G, Jiang Q, Fu P, Zhang Y, Jia Y, Yang X, Jiang H (2019) Comparative transcriptome analysis of gonads for the identification of sex-related genes in giant freshwater prawns (Macrobrachium rosenbergii) using RNA sequencing. Genes 10:E1035
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36:D480–D484
Kenny NJ, Sin YW, Shen X, Zhe Q, Wang W, Chan TF, Tobe SS, Shimeld SM, Chu KH, Hui JH (2014) Genomic sequence and experimental tractability of a new decapod shrimp model, Neocaridina denticulata. Mar Drugs 12:1419–1437
Kottler VA, Fadeev A, Weigel D, Dreyer C (2013) Pigment pattern formation in the guppy, Poecilia reticulata, involves the Kita and Csf1ra receptor tyrosine kinases. Genetics 194:631–646
Lee JH (2017) Quantitative approaches for investigating the spatial context of gene expression. Wiley Interdiscip Rev Syst Biol Med 9:e1369
Leu JH, Chen SH, Wang YB, Chen YC, Su SY, Lin CY, Ho JM, Lo CF (2011) A review of the major penaeid shrimp EST studies and the construction of a shrimp transcriptome database based on the ESTs from four penaeid shrimp. Mar Biotechnol 13:608–621
Levitt-Barmats Y, Yanai Z, Cohen TM, Shenkar N (2019) Life-history traits and ecological characteristics of the ornamental shrimp Neocaridina denticulata (De Haan, 1844), recently introduced into the freshwater systems of Israel. Aquat Invasions 14:684–702
Lynn Lamoreux M, Kelsh RN, Wakamatsu Y, Ozato K (2005) Pigment pattern formation in the medaka embryo. Pigment Cell Res 18:64–73
McMichael BK, Scherer KF, Franklin NC, Lee BS (2014) The RhoGAP activity of myosin IXB is critical for osteoclast podosome patterning, motility, and resorptive capacity. PLoS One 9:e87402
McNamara JC, Milograna SR (2015) Adaptive color change and the molecular endocrinology of pigment translocation in crustacean chromatophores. In: The Natural History of the Crustacea. 4:68–102
Milograna SR, Ribeiro MR, Baqui MM, McNamara JC (2014) Pigment granule translocation in red ovarian chromatophores from the palaemonid shrimp Macrobrachium olfersi (Weigmann, 1836): functional roles for the cytoskeleton and its molecular motors. Comp Biochem Physiol A Mol Integr Physiol 178:90–101
Milograna SR, Ribeiro MR, Bell FT, McNamara JC (2016) Pigment translocation in caridean shrimp chromatophores: receptor type, signal transduction, second messengers, and cross talk among multiple signaling cascades. J Exp Zool A Ecol Genet Physiol 325:565–580
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Mykles DL, Hui JH (2015) Neocaridina denticulata: a decapod crustacean model for functional genomics. Integr Comp Biol 55:891–897
Negisho T, Gemeda G, Du Laing G, Wakjira M, Janssens GPJ (2019) Diversity in micromineral distribution within the body of ornamental fish species. Biol Trace Elem Res. https://doi.org/10.1007/s12011-019-01983-1
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Neumann-Giesen C, Fernow I, Amaddii M, Tikkanen R (2007) Role of EGF-induced tyrosine phosphorylation of reggie-1/flotillin-2 in cell spreading and signaling to the actin cytoskeleton. J Cell Sci 120:395–406
Nguyen NH, Quinn J, Powell D, Elizur A, Thoa NP, Nocillado J, Lamont R, Remilton C, Knibb W (2014) Heritability for body colour and its genetic association with morphometric traits in Banana shrimp (Fenneropenaeus merguiensis). BMC Genet 15:132
Nong W, Chai ZYH, Jiang X, Qin J, Ma KY, Chan KM, Chan TF, Chow BKC, Kwan HS, Wong CKC, Qiu JW, Hui JHL, Chu KH (2020) A crustacean annotated transcriptome (CAT) database. BMC Genomics 21:32
Nur FAH, Christianus A (2013) Breeding and life cycle of Neocaridina denticulata sinensis (Kemp, 1918). Asian J Anim Vet Adv 8:108–115
Pan G, Yang J (2010) Analysis of microsatellite DNA markers reveals no genetic differentiation between wild and hatchery populations of Pacific threadfin in Hawaii. Int J Biol Sci 6:827–833
Parichy DM, Rawls JF, Pratt SJ, Whitfield TT, Johnson SL (1999) Zebrafish sparse corresponds to an orthologue of c-kit and is required for the morphogenesis of a subpopulation of melanocytes, but is not essential for hematopoiesis or primordial germ cell development. Development 126:3425–3436
Parichy DM, Mellgren EM, Rawls JF, Lopes SS, Kelsh RN, Johnson SL (2000) Mutational analysis of endothelin receptor b1 (rose) during neural crest and pigmentpattern development in the zebrafish Danio rerio. Dev Biol 227:294–306
Patoka J, Bláha M, Devetter M, Rylková K, Čadková Z, Kalous L (2015) Aquarium hitchhikers: attached commensals imported with freshwater shrimps via the pet trade. Biol Invasions 18:457–461
Pinnegar JK, Murray JM (2019) Understanding the United Kingdom marine aquarium trade - a mystery shopper study of species on sale. J Fish Biol 94:917–924
Riento K, Frick M, Schafer I, Nichols BJ (2009) Endocytosis of flotillin-1 and flotillin-2 is regulated by Fyn kinase. J Cell Sci 122:912–918
Sajeela KA, Gopalakrishnan A, Basheer VS, Mandal A, Bineesh KK, Grinson G, Gopakumar SD (2019) New insights from nuclear and mitochondrial markers on the genetic diversity and structure of the Indian white shrimp Fenneropenaeus indicus among the marginal seas in the Indian Ocean. Mol Phylogenet Evol 136:53–64
Shih HT, Cai Y (2007) Two new species of the land-locked freshwater shrimp genus, Neocaridina Kubo, 1938 (Decapoda: Caridea: Atyidae), from Taiwan, with notes on speciation on the island. Zool Stud 46:680–694
Sin YW, Kenny NJ, Qu Z, Chan KW, Chan KW, Cheong SP, Leung RW, Chan TF, Bendena WG, Chu KH, Tobe SS, Hui JH (2015) Identification of putative ecdysteroid and juvenile hormone pathway genes in the shrimp Neocaridina denticulata. Gen Comp Endocrinol 214:167–176
Sung HH, Chiu YW, Wang SY, Chen CM, Huang DJ (2014) Acute toxicity of mixture of acetaminophen and ibuprofen to green neon shrimp, Neocaridina denticulate. Environ Toxicol Pharmacol 38:8–13
Suzuki T, Kanou Y (2014) Two distinct arginine kinases in Neocaridina denticulate: psychrophilic and mesophilic enzymes. Int J Biol Macromol 67:433–438
Tang D, Shi X, Guo H, Bai Y, Shen C, Zhang Y, Wang Z (2020) Comparative transcriptome analysis of the gills of Procambarus clarkii provides novel insights into the immune-related mechanism of copper stress tolerance. Fish Shellfish Immunol 96:32–40
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Tuma MC, Gelfand VI (1999) Molecular mechanisms of pigment transport in melanophores. Pigment Cell Res 12:283–294
van der Salm AL, Metz JR, Wendelaar Bonga SE, Flik G (2005) Alpha-MSH, the melanocortin-1 Receptor and Background Adaptation in the Mozambique Tilapia, Oreochromis Mossambicus. Gen Comp Endocrinol 144:140–149
von Rintelen K, Page TJ, Cai Y, Roe K, Stelbrink B, Kuhajda BR, Iliffe TM, Hughes J, von Rintelen T (2012) Drawn to the dark side: a molecular phylogeny of freshwater shrimps (Crustacea: Decapoda: Caridea: Atyidae) reveals frequent cave invasions and challenges current taxonomic hypotheses. Mol Phylogenet Evol 63:82–96
Wade NM, Melville-Smith R, Degnan BM, Hall MR (2008) Control of shell colour changes in the lobster, Panulirus cygnus. J Exp Biol 211:1512–1519
Wade NM, Anderson M, Sellars MJ, Tume RK, Preston NP, Glencross BD (2012) Mechanisms of colour adaptation in the prawn Penaeus monodon. J Exp Biol 215:343–350
Wang J, Yu X, Zhao K, Zhang Y, Tong J, Peng Z (2012) Microsatellite development for an endangered bream Megalobrama pellegrini (Teleostei, Cyprinidae) using 454 sequencing. Int J Mol Sci 13:3009–3021
Wang W, Yang S, Wang C, Shi L, Guo H, Chan S (2017) Gill transcriptomes reveal involvement of cytoskeleton remodeling and immune defense in ammonia stress response in the banana shrimp Fenneropenaeus merguiensis. Fish Shellfish Immunol 71:319–328
Wang J, Ge Q, Li J, Gao H, Li J, Zhao F (2018) Identification of novel EST-SSR markers by transcriptome sequencing in ridgetail white prawn Exopalaemon carinicauda. Genes Genom 40:207–215
Wang F, Li S, Xiang J, Li F (2019) Transcriptome analysis reveals the activation of neuroendocrine-immune system in shrimp hemocytes at the early stage of WSSV infection. BMC Genomics 20:247
Wirth JA, Jensen KA, Post PL, Bement WM, Mooseker MS (1996) Human myosin-IXb, an unconventional myosin with a chimerin-like rho/rac GTPase-activating protein domain in its tail. J Cell Sci 109:653–661
Woolley TE, Maini PK, Gaffney EA (2014) Is pigment cell pattern formation in zebrafish a game of cops and robbers? Pigment Cell Melanoma Res 27:686–687
Wu JP, Li MH (2015) Inhibitory effects of pain relief drugs on neurological enzymes: implications on their potential neurotoxicity to aquatic animals. Environ Toxicol Pharmacol 39:898–905
Xu G, Du F, Nie Z, Xu P, Gu R (2014) Complete mitochondrial genome of Caridina nilotica gracilipes. Mitochondrial DNA 4:1–2
Yamada S, Tanaka Y, Sameshima M, Ito Y (1990) Pigmentation of prawn (Penaeus japonicus) with carotenoids: I. effect of dietary astaxanthin, β-carotene and canthaxanthin on pigmentation. Aquaculture 87:323–330
Yeh FC, Yang R, Boyle TJ, Ye Z, Xiyan JM (2000) POPGENE32, version 1.32. Microsoft Window-based freeware for population genetic analysis. University of Alberta, Edmonton, Canada
Yu YQ, Yang WJ, Yang JS (2014) The complete mitogenome of the Chinese swamp shrimp Neocaridina denticulata sinensis Kemp 1918 (Crustacea: Decapoda: Atyidae). Mitochondrial DNA 25:204–205
Yue GH, Chang A (2010) Molecular evidence for high frequency of multiple paternity in a freshwater shrimp species Caridina ensifera. PLoS One 5:e12721
Yue X, Nie Q, Xiao G, Liu B (2015) Transcriptome analysis of shell color-related genes in the clam Meretrix meretrix. Mar Biotechnol 17:364–374
Zeng HS, Ding SX, Wang J, Su YQ (2008) Characterization of eight polymorphic microsatellite loci for the giant grouper (Epinephelus lanceolatus Bloch). Mol Ecol Resour 8:805–807
Zhang J, Ma W, Song X, Lin Q, Gui JF, Mei J (2014) Characterization and development of EST-SSR markers derived from transcriptome of yellow catfish. Molecules 19:16402–16415
Zhao Y, Zhu X, Li Z, Xu W, Dong J, Wei H, Li Y, Li X (2019) Genetic diversity and structure of Chinese grass shrimp, Palaemonetes sinensis, inferred from transcriptome-derived microsatellite markers. BMC Genet 20:75
Acknowledgments
We are very grateful to Prof. CH Liou and YS Huang for their helpful comments on this work. We thank the members of the aquaculture team of our department for taking care of our ornamental shrimp breeding stocks.
Funding
This research is supported by the Fisheries Agency, Council of Agriculture, Taiwan, with grant number 104AS-16.2.6-FA-F1.
Author information
Authors and Affiliations
Contributions
C.W.H. was involved in planning and supervised the project. P.Y.C. and Y.F.W. contributed to the design and implementation of the research, to the analysis of the results, and to the writing of the manuscript. P.Y.C., Y.F.W., and W.R.C. processed the experimental data and performed the analysis. W.R.C. and Y.H.W. manufactured the samples. C.W.H., P.Y.C., Y.F.W., W.R.C., and Y.H.W. discussed the results and commented on the manuscript. C.W.H. finalized the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Huang, CW., Chu, PY., Wu, YF. et al. Identification of Functional SSR Markers in Freshwater Ornamental Shrimps Neocaridina denticulata Using Transcriptome Sequencing. Mar Biotechnol 22, 772–785 (2020). https://doi.org/10.1007/s10126-020-09979-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-020-09979-y