
Abstract
The European sea bass (Dicentrarchus labrax) is, along with the gilthead sea bream (Sparus aurata), one of the most extensively cultured species in European aquaculture productions. Massive mortalities may be caused by bacterial or viral infections in intensive aquaculture production. Evaluation of the efficacy of an oral vaccine against Vibrio anguillarum (Aquavac Vibrio Oral) in sea bass revealed specific immune gene expression profiles in the gut as well as protection of fish. In the present study, we performed RNA SEQ in two different tissues: the hind gut and the head kidney. For each tissue, one control sample (where a sample presents a pool of four to five individuals) and one sample after oral vaccine against V. anguillarum were submitted to 454 next-generation sequencing. In total, 269,043 sequences were obtained, 143,007 for head kidney and 125,036 for gut. The read lengths ranged from 40 to 706 bp with an average length of 348 bp. The total number of clustered sequences for head kidney is accounting to 49,089 (∼34 %) and for gut to 71,676 (∼57 %). Differential expression was detected for 496 transcripts in head kidney and for 336 in gut. The results not only enrich the present collection of expressed sequence tag sequences including rare transcripts like leukocyte immune-type receptors, cullin, or supervillin but also show the efficacy of oral vaccination against V. anguillarum.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Up until today, there are still a limited number of publications about vaccination in fish. Nevertheless, several viral fish vaccines as well as bacterial fish vaccines are available against serious viral and bacterial diseases. Examples of promising results for vaccination against viral infections have been obtained for rainbow trout (Oncorhynchus mykiss) reporting high levels of protection using DNA vaccines for viral hemorrhagic septicemia as well as for infectious hemorrhagic necrosis (Ambrose et al. 1991; Anderson et al. 1996; Corbeil et al. 1999; Kim et al. 2000; LaPatra et al. 2001). Furthermore, successful vaccination has been reported for the channel catfish (Ictalurus punctatus) against the channel catfish herpes virus type 1 (Nusbaum et al. 2002), for koi (Cyprinus carpio koi) against North American spring viremia of carp virus (Emmenegger and Kurath 2008) as well as for several salmonids against a range of different viral diseases (for a review, see Sommerset et al. 2005). Concerning bacterial diseases, vaccines have been reported for salmonids, channel catfish, carp, Japanese flounder, eel, rainbow trout, tilapia, sea bream as well as sea bass (for a review, see Sommerset et al. 2005). For sea bass, vaccination against Pasteurellosis (Photobacterium damsela subspecies piscicida) is available as well as against Vibrio anguillarum providing protection to some degree. However, the nature of cellular immune response against V. anguillarum is largely unknown. It has been shown that down-regulation of apoptotic caspases in close linkage to the inhibition of reactive oxygen intermediates generation in professional phagocytes perform as a combined virulence mechanism.
The fish immune systems are primitive when compared to mammals (Lieschke and Trede 2009). Fish, however, are the first group of animals with the basic aspects of the immune system in higher vertebrates. Considering that the number of fish species is significantly higher than the number of mammals and that the evolutionary distance between species is greater in teleosts than in Mammalia, it is obvious that efficient general vaccination may not be achieved. Identification and characterization of transcripts involved in the cellular immune response and accumulation of knowledge of the immune system in different fish species as well as the study of pathogen and vaccine-induced immunity are essential in order to enhance the development of a cost effective but efficient vaccine for important commercial marine fish species like the European sea bass. The European sea bass (Dicentrarchus labrax) is, along with the gilthead sea bream (Sparus aurata), one of the most extensively cultured species in European aquaculture productions. Bacterial or viral infection may initiate massive mortalities in intensive aquaculture production. Specific immune gene expression profiles in the gut as well as protection of fish demonstrated the efficacy of an oral vaccine against V. anguillarum (Aquavac Vibrio Oral) in sea bass. Next-generation sequencing (NGS) provides great potential to unravel further genes involved in immune response as well as to detect differential gene expression. Detection of differential expression by counting transcripts has been successfully shown in several previous studies (e.g., Sarropoulou et al. 2009). In the present study, we performed RNA SEQ (Wang et al. 2009) in two different tissues: gut and head kidney (HK). For each tissue, one control sample (where a sample presents a pool of four to five individuals) and one sample after oral vaccine against V. anguillarum were submitted to 454 next-generation sequencing.
Materials and Methods
Animals
Healthy juvenile specimens of European sea bass (D. labrax L.) (70 g mean initial weight) from the Spanish Oceanographic Institute (Mazarron, Murcia) were reared in 14 m3 running seawater aquaria with an initial density of 200 fish/tank. Temperature and photoperiod followed a natural pattern over the whole experimental period. All animals were handled in compliance with the Guidelines of the European Union Council (86/609/EU) and the Bioethical Committee of the University of Murcia (Spain) for the use of laboratory animals.
Feeding and Vaccination
Fish were fed at a feeding rate of 1.5 % of fish biomass with a commercial pellet diet (Trouvit, Spain) alone or supplemented over 10 days with a commercial oral vaccine designed for the active immunization of fish to reduce mortality due to vibriosis caused by Listonella (V.) anguillarum and Vibrio ordalii (Aquavac Vibrio Oral, ISPAH). A primary vaccine at the starting of the trial and a booster 4 months later were administered, mixed with food pellets, following the manufacturer’s suggested protocol. The scheme implied 5 days administration with 0.02 ml/vaccine per day followed by 5 days without vaccine and again 5 days vaccine with the same dose as in the beginning to accomplish with 0.2 ml per fish. The relative percentage of survival achieved by the vaccine at 30 and 80 days post-booster was 57 and 33 %, respectively.
Tissue Collection and RNA Isolation
On day 156 (1 day after booster and 135 after primary vaccination), fish were instantly killed with an overdose of the anesthetic tricaine methane sulfonate (MS222 Sigma). Immediately after the fish died, HK and hind gut (named as gut hereafter) were removed from ten individual (five individuals per each tissue/treatment) for RNA extraction, collected in RNAlater (Sigma-Aldrich) and stored at −80 °C (Fig. 1).

Sampling diagram showing the timing and administration of vaccine feed as well as the sampling point used for sequencing
Total RNA from four tissues (HK, after oral vaccination and control, gut after oral vaccination and control) was obtained using the NucleoSplin RNA II extraction kit (Machinery Nagel, Dueren, Germany). Quality run at the DNA analyzer (Agilent 2100 Bioanalyzer, Palo Alto, CA, USA) was performed, and quantity was measured using NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA).
454 cDNA Library Construction and 454 GS-Titanium Sequencing
For cDNA library construction, total RNA was fragmented following the instructions of 454 cDNA rapid library protocol with few alternations. In brief, 2-μg total RNA was fragmented by adding 2-μl fragmentation solution in a total volume of 18 μl, vortexed and incubated at 72 °C for 30 s. Precipitation was performed at −20 °C overnight in a total volume of 500 μl with 50 μl NaOAc, 2.5 ml EtOH, and 1 μl glycogen (Invitrogen/VWR, Tromsø, Norway). Synthesis of first-strand cDNA was performed using oligonucleotide primer 5′-TTTTTTCTTGTTTTCTTTTCTTV-3′, and cDNA library protocol of GSFLX45 was adapted accordingly (Supplemental figure 1). Second-strand synthesis as well as library preparation was constructed following the instructions of GSFLX cDNA rapid library protocol. Quantification of the library was assessed by using the Quantiflur ST Fluorometer (SB Biotechnology Suppliers S.A), and fragment size distribution was determined by analyzing 1 μl of the samples on the BioAnalyzer (Agilent 2100). Next-generation sequencing was performed according to GSFLX Titanium protocols. All sequences were submitted to the NCBI SRA database under the accession number SRA050000.
Bioinformatic Analysis
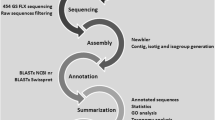
For bioinformatic analysis purposes, sff files were converted to fasta files and sequences were trimmed and clustered using free available software analysis tools SeqClean (http://wwwtigrorg/tdb/tgi/software/) and cdHit (Li and Godzik 2006; Huang et al. 2010), respectively. Transcripts represented abundantly in one of the libraries were selected for in silico expression analysis after (Stekel et al. 2000). In brief, all clusters were submitted to a log likelihood ratio statistics which tends asymptotically to a χ 2 distribution described by Stekel et al. (2000). It is based on a single statistical test to describe the extent to which a gene is differentially expressed between libraries. This method permits in any number of libraries to identify differential expressed genes. For evaluation purposes, a second method for differential detection, the DEGseq package for R (Wang et al. 2010), was applied. This package is a free R package to detect differentially expressed genes from RNA SEQ data. It is used to identify differential expression of genes between two samples with or without replicates.
Results
454-Sequence Analysis
After trimming of sequences, a total of 269,043 sequences were obtained: for head kidney 143,007 and for gut 125,036. The read lengths ranged from 40 to 706 bp with an average length of 348 bp (Table 1). The total number of clustered sequences for head kidney is accounting to 49,089 (∼34 %) and for gut to 71,676 (∼57 %). Cluster distribution for both tissues is shown in supplemental figure 2.
Expression Analysis
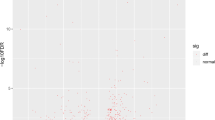
For expression analysis, control and treated libraries of each tissue were compared to each other by looking at the abundance of a transcript. Therefore, the extent to which a transcript is expressed differentially between the two libraries is tested by a log likelihood ratio statistics described in Stekel et al. (2000). Transcripts are plotted as a function of the obtained values of the test statistic R (Fig. 2). It can be seen that for both tissues head kidney and gut, the number of genes decreases exponentially for R ≤ 5. The slope for this region is −0.90 and −0.95 for head kidney and gut, respectively; both are not significantly different from −1 at 5 % significance. Transcripts with R ≥ 5 are accounting for head kidney to 496 and for gut to 336. Figures 3 and 4 present a set of transcripts showing significant regulation between control and vaccinated tissues. Using the DEGseq package for R revealed for head kidney 413 differential expressed genes and for gut 296 using a threshold of p = 0.001 (Fig. 5).

The number of genes for a given value or the test statistic R is plotted as a function of R. The data falling within 1 < R < 5 are decrees exponential curve and decreasing exponentially with R. The slope is −0.95251 with significance at the 5 % level of 0.013 and is therefore not significantly different from −1 at 5 % significance. When R > 5, the number of genes is above this exponential curve

A set of genes showing significant differential expression between vaccinated and control tissue in head kidney. Transcripts were counted within each cluster obtained and R values were calculated for statistical significance

A set of genes showing significant differential expression between vaccinated and control tissue in gut. Transcripts were counted within each cluster obtained and R values were calculated for statistical significance

MA plot of head kidney and gut using DEGseq package for R. Differential expressed genes are the red points in the MA plot
Annotation and Gene Ontology
Sequence similarity searches were performed against the NCBI nonredundant nucleotide database (BlastN) as well as against the NCBI protein database (BlastX). For head kidney, 27,614 (56 %) were assigned to BlastN hits whereas 16,073 (∼33 %) sequences were successfully assigned to BlastX hits. Out of the successfully assigned BlastX hits, 57 % were mapped using Blast2Go analysis tool and 45 % were successfully annotated. Analysis was performed for all transcripts of both treatments: control and vaccinated head kidney as well as separately. GO analysis did not differ between the two treatments (data not shown). Results of assigning GO numbers of molecular function at level 2 is illustrated in Fig. 6a.

Gene Ontology analysis of clustered sequences using molecular functions at level 2. Analysis was performed with Blast2Go software packagea. A GO analysis for transcripts obtained from head kidney. B GO analysis for transcripts obtained from gut
Concerning gut, 30,167 (42 %) were assigned to BlastN hits whereas 14,869 (∼21 %) sequences were successfully assigned to BlastX hits. Out of the successfully assigned BlastX hits, 74 % were mapped using Blast2Go analysis tool and 59 % were successfully annotated. Analysis was performed for all transcripts of both treatments: control and vaccinated head kidney as well as separately. GO analysis did not differ between the two treatments (data not shown). Results of assigning GO numbers of molecular function at level 2 is illustrated in Fig. 6b. In head kidney, three additional GO categories in comparison with gut were observed: chemoattractant activity, proteasome regulatory activity, and metallochaperone activity. In both tissues, the categories binding and catalytic activity were found to be the most abundant categories. All Blast results are available in supplemental Tables 1 and 2 for head kidney and gut, respectively.
Novel Sequences
BlastN hits showing already annotated sequences of the European sea bass accounted in head kidney to 5,614 and in gut to 10, 826. Almost 50 % 2,339 and 4,674 sequences, respectively, resulted in a positive hit with the description “D. labrax chromosome sequence corresponding to linkage group top complete sequence.”
Besides the R values, indications of genes are shown for which transcripts were found either in control or vaccinated tissues, e.g., calpastin. Other transcripts were found in different clusters, but same Blast hit like, e.g., nicotinamide riboside kinase 2. Here 24 clusters were found to encode for nicotinamide riboside kinase 2 with in total 81 sequences. Alignment of the 24 clusters is showing significant differences between the sequences. In the case of switch-associated protein 70, nine sequences were grouped into seven different clusters. Interestingly, out of the seven clusters, two clusters were detected only in vaccinated fish whereas seven transcripts apparently encoding for the same genes but were found to be in different clusters. Nine different transcripts with the same Blast hit were found for the gene supervillin. Seven of the variants are expressed only in the vaccinated sample and two variants in the control. Table 2 shows further examples of possible isoforms or paralogs transcripts identified in the present study.
Discussion
NGS or massively parallel pyrosequencing experienced a very fast development in view of sequence length, high throughput, and accuracy. A broad range of next-generation sequencing technologies like SOLid, Helicos BioSciences’ Heliscope, PacBio, Illumina’s Genome Analyzer, IonTorrent, and 454 GS Roche Life Sciences sequencing systems are available today. Differences between NGS instruments are mainly in sequence length, throughput, and cost (for a review, see Glenn 2011 and Loman et al. 2012). For non-model species for which no reference transcriptome is available, read length is yet, primarily for annotation purposes, of importance. In the present study, the 454 GSFLX Titanium sequencing platform was applied as it was aimed to detect differential expression in the European sea bass (D. labrax), a non-model species of commercial interest. The average read length of 348 bp obtained in the present work is in line with the expectations of 454 GS Titanium sequencing platform as well as with other transcriptome sequencing approaches (e.g., Droege and Hill 2008; Salem et al. 2010; Yufera et al. 2011). High-throughput sequencing has been applied mainly to sequence whole genomes and high fold amplicon sequencing in order to detect rare allele variants. Concerning transcriptome sequencing, most studies for non-model species are aiming to obtain first insights into the whole transcriptome rather than looking at differential expression. In order to detect differential expression, each transcript has to be counted once. High-throughput gene expression studies were performed mainly by using microarray technology. However, this technique beside several advantages implements also serious of disadvantages. Similar sequences like the members of a multigene family or paralogs may not be detected as well as the complexity of hybridization behavior. In addition, the expression analysis only of the plotted transcripts is analyzed. Expressed sequence tag (EST) sequencing is promising but expensive method to measure gene expression. Using NGS technologies provides a cost-effective and fast access to get hold of EST sequences. In this line, Torres et al. (2008) showed that 3′ UTR sequencing of randomly sheared cDNA molecules can be applied for expression profiling. Nevertheless, most studies are showing relative abundance of transcripts performing random transcriptome sequencing followed by transcriptome assembling. In the present study, oligo-d(T) specially designed for 454 sequencing was used for poly(A) tagging; hence, the 3′ UTR of each transcript was sequence only once (see supplement Fig. 1). Consequently, no assembly had to be performed allowing the identification of similar transcripts like isoforms or paralogs on the one hand, but on the other hand, the construction of full-length cDNA was hindered as the maximum read length of the 454 GS FLX run was 729 bp. In total, 92,647,912 bp of DNA sequence was obtained. The genome of the European sea bass is estimated to be around 600 Mb from which ∼1 % is assumed to correspond to mRNA (Salem et al. 2010). Thus, obtained sequences in the present study provide a 15× depth coverage of the sea bass transcriptome.
Sequences have been clustered for each tissue separately, and the number of clustered sequences for gut is accounting to 71,676 (∼57 %) and for head kidney to 49,089 (∼34 %). Interestingly in other studies where sequences were assembled but not clustered as random primers were used for cDNA synthesis, the percentage of unique sequences was about 30 % in the rainbow trout (Salem et al. 2010), 42 % in the lake sturgeon (Hale et al. 2009), and 6 % in the gilthead sea bream after first assembling and then clustering (Yufera et al. 2011).
Functional annotation of clustered sequences was carried out by BlastX as well as BlastN search. The top-hit species distribution positioned Oreochromis niloticus at first place indicating the high amount of sequence information in this species.
Looking at the GO category molecular function at level 2, differences between head kidney and gut are shown. The categories metallochaperone activity, proteasome regulator activity, as well as chemoattractant activity were not represented in the gut transcriptome analysis of the GO molecular function analysis. As in most teleost tissue studies up until today (e.g., Sarropoulou et al. 2009; Salem et al. 2010; Yufera et al. 2011), the category “binding proteins” was the most abundant category.
Differential expression was detected by using the test statistic as described in (Stekel et al. 2000). This test has been developed to compare the abundance of a transcript in any number of cDNA libraries taking into account the number of sequences obtained in each of the library. In the present study, transcripts with R ≥ 5 are statistical significantly different expressed between vaccinated and control tissue. Transcripts in head kidney with R ≥ 5 are accounting to 496 and transcripts in gut with R ≥ 5 are accounting to 336. Figures 3 and 4 present a set of transcripts showing significant regulation between control and vaccinated tissues. MRNAs with low expression levels were also detected like LITR which can be attributed to the depth of NGS sequencing technologies. In addition, several clustered sequences were found with similar gene annotation reflecting the power of 3′ UTR sequencing for isoform or paralog identifications.
An interesting observation is that the above statistical tests were unable to find significant differences for some immune-related genes identified mainly in vaccinated animals, probably because of the low abundance of these transcripts and the presence of several isoforms in most of them (Tables 3 and 4). However, some of these genes might be relevant from an immunological point of view and might be involved in the protection confers by oral vaccines. Among them, several IFN-stimulated genes, TNF receptors and signaling molecules, purinoreceptors, and histamine receptors are particularly interesting. In addition, intriguing is the presence of several genes encoding retrotransposon members in vaccinated fish. Although it is difficult to anticipate the role of these molecules in vaccination, our data pave the way for future studies aimed to clarify the role of transposable elements in mucosal immunity and the signaling mechanisms involved in the induction of these genes upon a antigenic challenge.
Conclusion
In conclusion, the head kidney and the gut transcriptome of control and vaccinated fish were successfully sequenced using next-generation sequencing technology. Differential expression was assessed by using a modified oligo-d(T) primer for 3′ UTR tagging unraveling a set of potentially differential expressed genes between vaccinated and control tissues as well as rare transcripts as a result of deep sequencing. In addition, candidate transcripts pinpointing to different isoforms or paralogs were detected due to targeted 3′ UTR sequencing.
References
Ambrose MW, Wyde PR, Ewasyshyn M, Bonneau AM, Caplan B, Meyer HL, Klein M (1991) Evaluation of the immunogenicity and protective efficacy of a candidate parainfluenza virus type 3 subunit vaccine in cotton rats. Vaccine 9:505–511
Anderson ED, Mourich DV, Fahrenkrug SC, Lapatra S, Shepherd J, Leong JA (1996) Genetic immunization of rainbow trout (Oncorhynchus mykiss) against infectious hematopoietic necrosis virus. Mol Mar Biol Biotechnol 5:114–122
Corbeil S, Lapatra SE, Anderson ED, Jones J, Vincent B, Hsu YL, Kurath G (1999) Evaluation of the protective immunogenicity of the N, P, M, NV and G proteins of infectious hematopoietic necrosis virus in rainbow trout Oncorhynchus mykiss using DNA vaccines. Dis Aquat Organ 39:29–36
Droege M, Hill B (2008) The Genome Sequencer FLX System—longer reads, more applications, straight forward bioinformatics and more complete data sets. J Biotechnol 136:3–10
Emmenegger EJ, Kurath G (2008) DNA vaccine protects ornamental koi (Cyprinus carpio koi) against North American spring viremia of carp virus. Vaccine 26:6415–6421
Glenn TC (2011) Field guide to next-generation DNA sequencers. Mol Ecol Resour 11:759–769
Hale MC, Mccormick CR, Jackson JR, Dewoody JA (2009) Next-generation pyrosequencing of gonad transcriptomes in the polyploid lake sturgeon (Acipenser fulvescens): the relative merits of normalization and rarefaction in gene discovery. BMC Genomics 10:203
Huang Y, Niu B, Gao Y, Fu L, Li W (2010) CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26:680–682
Kim CH, Johnson MC, Drennan JD, Simon BE, Thomann E, Leong JA (2000) DNA vaccines encoding viral glycoproteins induce nonspecific immunity and Mx protein synthesis in fish. J Virol 74:7048–7054
Lapatra SE, Corbeil S, Jones GR, Shewmaker WD, Lorenzen N, Anderson ED, Kurath G (2001) Protection of rainbow trout against infectious hematopoietic necrosis virus four days after specific or semi-specific DNA vaccination. Vaccine 19:4011–4019
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659
Lieschke, GJ, Trede, NS (2009) Fish immunology. Curr Biol 19:R678–82
Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ (2012) Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. doi:10.1038/nbt.2198
Nusbaum KE, Smith BF, Deinnocentes P, Bird RC (2002) Protective immunity induced by DNA vaccination of channel catfish with early and late transcripts of the channel catfish herpesvirus (IHV-1). Vet Immunol Immunopathol 84:151–168
Salem M, Rexroad CE 3rd, Wang J, Thorgaard GH, Yao J (2010) Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics 11:564
Sarropoulou E, Sepulcre P, Poisa-Beiro L, Mulero V, Meseguer J, Figueras A, Novoa B, Terzoglou V, Reinhardt R, Magoulas A, Kotoulas G (2009) Profiling of infection specific mRNA transcripts of the European seabass Dicentrarchus labrax. BMC Genomics 10:157
Sommerset I, Krossoy B, Biering E, Frost P (2005) Vaccines for fish in aquaculture. Expert Rev Vaccines 4:89–101
Stekel DJ, Git Y, Falciani F (2000) The comparison of gene expression from multiple cDNA libraries. Genome Res 10:2055–2061
Torres TT, Metta M, Ottenwalder B, Schlotterer C (2008) Gene expression profiling by massively parallel sequencing. Genome Res 18:172–177
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wang L, Feng Z, Wang X, Zhang X (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138
Yufera M, Halm S, Beltran S, Fuste B, Planas JV, Martinez-Rodriguez G (2011) Transcriptomic characterization of the larval stage in gilthead seabream (Sparus aurata) by 454 pyrosequencing. Mar Biotechnol (NY) 14:423–435
Acknowledgments
We thank Inma Fuentes for her excellent technical assistance. This work was supported by the Consejería de Universidades, Empresa e Investigación de la Región de Murcia (Spain) (grant BIO-GAN 07/01-0001 to V.M.), and Culmarex, S.A. (Spain).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Fig7
Supplemental figure 1 Schematic illustration of RNA fragmentation and first-strand cDNA synthesis using oligonucleotide primer 5′-TTTTTTCTTGTTTTCTTTTCTTV-3′. (JPEG 716 kb)
Fig8
Supplemental figure 2 Cluster size distribution in European sea bass head kidney and gut. On the x-axes, the amount of sequences is given whereas on the y-axes the obtained cluster size. (JPEG 131 kb)
10126_2012_9466_MOESM1_ESM.csv
Supplemental Table 1 BlastN results of clustered sequences obtained from control and vaccinated D. labrax head kidney tissue. (CSV 6378 kb)
10126_2012_9466_MOESM2_ESM.csv
Supplemental Table 2 BlastX results of clustered sequences obtained from control and vaccinated D. labrax head kidney tissue. (CSV 3108 kb)
10126_2012_9466_MOESM3_ESM.csv
Supplemental Table 3 BlastN results of clustered sequences obtained from control and vaccinated D. labrax gut tissue. (CSV 7138 kb)
10126_2012_9466_MOESM4_ESM.csv
Supplemental Table 4 BlastX results of clustered sequences obtained from control and vaccinated D. labrax gut tissue. (CSV 2726 kb)
10126_2012_9466_MOESM6_ESM.csv
Supplemental Table 6 Results of pathway maps from KEGG obtained from vaccinated D. labrax head kidney tissue. (CSV 35 kb)
Rights and permissions
About this article
Cite this article
Sarropoulou, E., Galindo-Villegas, J., García-Alcázar, A. et al. Characterization of European Sea Bass Transcripts by RNA SEQ After Oral Vaccine Against V. anguillarum . Mar Biotechnol 14, 634–642 (2012). https://doi.org/10.1007/s10126-012-9466-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-012-9466-z