
Abstract
The extracellular phytase in the supernatant of cell culture of the marine yeast Kodamaea ohmeri BG3 was purified to homogeneity with a 7.2-fold increase in specific phytase activity as compared to that in the supernatant by ammonium sulfate fractionation, gel filtration chromatography (Sephadex™ G-75), and anion-exchange chromatography (DEAE Sepharose Fast Flow Anion-Exchange). According to the data from sodium dodecyl sulfate-polyacrylamide gel electrophoresis, the molecular mass of the purified enzyme was estimated to be 98.2 kDa while the molecular mass of the purified enzyme was estimated to be 92.9 kDa and the enzyme was shown to be a monomer according to the results of gel filtration chromatography. The optimal pH and temperature of the purified enzyme were 5.0 and 65°C, respectively. The enzyme was stimulated by Mn2+, Ca2+, K+, Li+, Na+, Ba2+, Mg2+ and Co2+ (at a concentrations of 5.0 mM), but it was inhibited by Cu2+, Hg2+, Fe2+, Fe3+, Ag+, and Zn2+ (at a concentration of 5.0 mM). The enzyme was also inhibited by phenylmethylsulfonyl fluoride (PMSF), iodoacetic acid (at a concentration of 1.0 mM), and phenylgloxal hydrate (at a concentration of 5.0 mM), and not inhibited by EDTA and 1,10-phenanthroline (at concentrations of 1.0 mM and 5.0 mM). The K m, V max, and K cat values of the purified enzyme for phytate were 1.45 mM, 0.083 μmol/ml · min, and 0.93 s-1, respectively.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phytase (myo-inositol hexakisphosphate phosphohydrolase, EC 3.1.3.8) catalyzes the release of phosphate from phytate (myo-inositol hexakiphosphate), which is the principle type of phosphorus present in cereal grains, legumes, and oilseeds (Pandey et al. 2001). Therefore, phytase can be incorporated into commercial poultry, swine, and fish diets and has a wide range of applications in animal and human nutrition as it can reduce phosphorus excretion of monogastric animals by replacing inorganic phosphates in the animal diet; contribute significantly toward environmental protection; and lead to improved availability of minerals, trace elements, amino acids, and energy (Vats and Banerjee 2004; Haefner et al. 2005).
In recent decades, the global maricultural industry, including shrimp, bivalve, seaweed, and sea cucumber farming, has been developing rapidly as a result of a high demand for sea food. Non-eco-friendly management practices that are very common in intensive modern farming systems such as discharges of waste manures and waste water from treatment plants appear to be associated with phosphorus pollution in marine environment. Therefore, phytase also can be incorporated into the maricultural feed because phytase is lacking in the guts of marine farming animals (Chi et al. 2006).
Microorganisms are the best sources for commercial production of phytases because of their easy cultivation and high yields of the enzyme. At present, all phytase preparations authorized in the EU as feed additives are produced by recombinant strains of filamentous fungi and the expressed phytase genes are of fungal origin and originate in two cases from the genus Aspergillus (Haefner et al. 2005). In the last decade, phytate-degrading enzymes of terrestrial yeasts such as Schwanniomyces castellii (Pandey et al. 2001), Schwanniomyces occidentalis (Nakamura et al. 1999), Pichia anomala (Vohara and Satyanarayana 2004), Arxula adeninivorans (Sano et al. 1999; Kaur et al. 2007), Hansenula polymorph (Mayer et al. 1999), and Rhodotorula gracilis (Bindu et al. 1998) also have received increasing attention as they can be easily incorporated into feed diets and are rich in nutrients. So far, extracellular phytase from A. adeninivorans (Sano et al. 1999), Pichia anomala (Han and Lei 1999), Saccharomyces cerevisiae (Han et al. 1999), and S. occidentalis (Segueilha et al. 1992) has been purified and characterized and the Apho1p gene for secretory acid phosphatase from A. adeninivorans strain LS3 has been cloned (Kaur et al. 2007). It also has been reported that the gene encoding phytase from Debaryomyces castellii CBS 2923 has been cloned, overexpressed, and the recombinant enzyme has been characterized. However, little is known about phytase from marine yeasts.
In our recent studies (Hirimuthugoda et al. 2006), we found that phytase activity produced by Kodamaea ohmeri BG3 isolated from the gut of the marine fish (Hexagrammes otakii) reached greater than 557.9 mU/ml under the optimal conditions. Our purpose in the present study was to purify and characterize the extracellular phytase produced by the marine yeast strain. This is the first report on phytase from marine yeasts.
Materials and Methods
Marine Yeast Strain
The marine yeast strain BG3, identified to be a strain of Kodamaea ohmeri, was isolated from gut of marine fish (Hexagrammes otakii) (Hirimuthugoda et al. 2006) and maintained on YPD medium containing 1.0% yeast extract, 2.0% polypeptone, and 2.0% glucose at 4°C.
Phytase Production
One loop of the cells of the yeast strain was transferred to 50 ml of YPD medium prepared with distilled water in a 250-ml flask and aerobically cultivated for 24 h. The cell culture (1.0 ml, OD600nm = 10.0) was centrifuged at 6225 g and 4°C for 5 min and washed three times with sterile saline water. The washed cells were transferred to 50 ml of the production medium, which contained 1.0% oat, 3.0% glucose, 1.5% ammonium sulfate, 2.0% NaCl, pH 5.0 and grown by shaking at 170 rpm and 28°C for 3 days.
Phyatse Assay
One mililiter of the cell culture was centrifuged at 6225 g for 10 min. The supernatant obtained was used as the crude extracellular phytase preparation. The phytase activity was assayed as follow: 0.8 ml of sodium phytate solution (5.0 mM sodium phytate in 0.2 M sodium acetate, pH 5.0) was preincubated at 60°C for 5 min and 0.2 ml of the extracellular phytase preparation was added and mixed well. The mixture was incubated 60°C for 30 min. The reaction was stopped by addition of 1.0 ml of 5.0% trichloroacetic acid (TCA). Inorganic phosphate liberated was quantitatively determined spectrophotometrically at 700 nm via the ammonium molybdate method (Chi et al. 1999). One unit of phytase activity was defined as the amount of enzyme causing the release of 1.0 μmol of inorganic phosphate/min under the assay conditions. Protein concentration was measured by the method of Bradford, and bovine serum albumin served as the standard (Bradford 1976).
Enzyme Purification
Enzyme purification was carried out at 4°C. Three liters of the culture grown for 3 days were used as the starting material for phytase purification. After removal of the cells by centrifugation at 14,006 g for 20 min, proteins in the resulting supernatant were precipitated by ammonium sulfate and dialyzed against 0.2 M acetate buffer (pH 5.0). The dialyzed protein solution was then applied to a Sephadex™ G-75 column (medium grade; Pharmacia 2.5 × 100 cm) and the column was eluted with 0.2 M acetate buffer (pH 5.0) by using ÄKTA™ prime with Hitrap™ (Amersham, Biosciences, Sweden). At a flow rate of 0.5 ml/min, 3.0-ml fractions were collected. The phytase-positive fractions were combined and dialyzed in 20 mM Tris-HCl buffer (pH 7.5) overnight. The dialyzed phytase-positive elute was applied to a DEAE Sepharose Fast Flow anion-exchange column (2.5 × 30 cm) that had been equilibrated with 20 mM Tris-HCI buffer (pH 7.5) and the column was washed with the same buffer for 2 h at a flow rate of 1 ml/min. The bound proteins were then eluted with a linear gradient of NaCl solution in the range of 0 to 1.0 M in the equilibrating buffer. The phytase-positive fractions were concentrated by filtration through an AmiconYM3 (MW cut-off 10,000) membrane.
Estimation of Molecular Mass of the Purified Phytase by Gel Filtration Chromatography
The blue dextran (the final concentration was 2.0 mg/ml) was dissolved in equilibration buffer containing 5% glycerol. A 2.0-ml aliquot of the solution was applied to a 100 cm × 2.5 cm Sephadex™ G-200 (Pharmacia) column that had been equilibrated with the buffer of 50 mM Tris-HCl and 100 mM KCl (pH 7.5) and the flow rate was 0.3 ml/min. The void volume, which equaled the volume of effluent collected from the point of sample application to the center of the effluent peak, was determined. The individual protein standards (albumin 66.0 kDa, 10 mg/ml; alcohol dehydrogenase 150.0 kDa, 5 mg/ml; β-amylase 200.0 kDa, 4 mg/ml; carbonic anhydrase 29.0 kDa, 3 mg/ml; cytochrome c 12.4 kDa, 2 mg/ml) were dissolved in equilibration buffer containing 5% glycerol. A 2.0-ml aliquot of the solution of protein standards was applied to a 100 cm × 2.5 cm Sephadex™ G-200 (Pharmacia) column that had been equilibrated with the buffer of 50 mM Tris-HCl and 100 mM KCl (pH 7.5) and the flow rate was 0.3 ml/min. The V e for the protein standards was obtained by measuring the volume of effluent collected from the point of sample application to the center of the effluent peak. Calibration curve (molecular mass vs. V e/V o) was made according to V o and V e values obtained above and molecular mass of the individual protein standards. The V e of the purified phytase was determined using the same methods applied to the standards and the V e/V o and its molecular mass from the standard curve were calculated.
Gel Electrophoresis
The purity and molecular mass of phytase in the concentrated fractions showing the activity were analyzed in noncontinuous denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; Laemmli 1970) with a two-dimensional electrophoresis system (Amersham, Biosciences, Sweden) and stained by Coomassie brilliant blue R-250 (George and Diwan 1983). The molecular mass standards for SDS-PAGE comprised β-galactosidase (116 kDa), bovine serum albumin (66.2 kDa), ovalbumin (45 kDa), lactate dehydrogenase (35 kDa), rease Bsp981 (25 kDa), and β-lactoglobulin (18.4 kDa).
Cloning of the Full Length of the Gene Encoding Phytase in Kodamaea ohmeri BG3
Genomic DNA from Kodamaea ohmeri BG3 was isolated according to the method described by Adams et al. (1998). To isolate the consensus sequence of partial gene encoding phytase from Kodamaea ohmeri BG3, one set of degenerated primers (PP: 5′-AGACAYGGTGARAGRTAYCC-3′ and PR: 5′-ACAWGAGAAWCCWGGWCC) designed based on the sequence information of phytase genes from D. castellii (ABN04184), A. adeninivorans (AM231307), Pichia guilliermondii (CAL69849) were used for polymerase chain reaction (PCR) with genomic DNA of Kodamaea ohmeri BG3 as template. The reaction system (25 μl) was composed of 2.5 μl of 10 × buffer, 2.0 μl (2.5 mM) of dNTP, 1.0 μl (100 μM) of PP, 1.0 μl (100 μM) of PR, 0.5 μl of Taq DNA polymerase, 1.0 μl (10.0 ng/μl) of template DNA, and 17.0 μl of H2O. The conditions for the PCR amplification were as follows: initial denaturation at 94°C for 10 min, denaturation at 94°C for 1 min, annealing temperature at 53°C for 1 min, extension at 72°C for 2 min, final extension at 72°C for 10 min. PCR was run for 32 cycles and the PCR cycler was GeneAmp PCR System 2400 (PerkinElmer, Waltham, MA). The PCR products were cloned into pGEM-T easy vector (Promega, Madison, WI) and sequenced. According to the sequence, the full length sequence of the gene encoding phytase in Kodamaea ohmeri BG3 was cloned by using SMART™ RACE cDNA Amplification Kit (Clontech, Mountain View, CA). The amino acid sequence of the cloned DNA sequence was deduced by using the software offered in NCBI. Multiple alignments of the amino acid sequence were produced using DNAMAN 6.0.
Effects of pH and Temperature on Phytase Activity and Stability
The effect of pH on the purified phytase activity was determined by incubating the purified enzyme between pH 3.0 and 9.0 using the standard assay conditions. The buffers used were 0.2 M acetate buffer (pH 3.0 to 6.0) and 0.2 M Na2B407·10 H2O–H3BO3 buffer (pH 7.0 to 10.0). The pH stability was tested via 6-h preincubation of the purified enzyme in appropriate buffers that had the same ionic concentrations at different pH values ranging from 3.0 to 10.0 at 0°C. The remaining activities of phytase were measured immediately after this treatment with the standard method as mentioned earlier.
The optimal temperature for activity of the enzyme was determined at 30°C, 35°C, 40°C, 45°C, 50°C, 55°C, 60°C, 65°C, 70°C, 75°C, and 80°C in the same buffer as described in the preceding text. Temperature stability of the purified enzyme was tested by preincubating the enzyme at different temperatures (20°C, 40°C, 60°C, 65°C, and 70°C) for 1 h, the residual activity was measured immediately as described in the preceding text. Here, preincubated sample at 0°C was used a reference to calculate the residual activity.
Effects of Different Metal Ions and Enzyme Inhibitors on Phytase Activity
To examine effects of different metal ions on phytase activity, an enzyme assay was performed for 1 h in the reaction mixture as described in the preceding text with various metal ions at a final concentrations of 1.0 mM and 5.0 mM. The activity assayed in the absence of metal ions was defined as the control. The metal ions tested include Zn2+, Cu2+, Mg2+, Fe3+, Ca2+, K+, Mn2+, Hg2+, Li+, Fe2+, Ag+ , Na+, Ba2+, and Co2+.
The effects of enzyme inhibitors (EDTA, PMSF, 1,10-phenanthroline, iodoacetic acid, and phenylgloxal hydrate at a final concentration of 1.0 and 5.0 mM, respectively) on the phytase activity were measured in the reaction mixture as described earlier. The purified enzyme was preincubated with the respective compound for 1 h at 0°C, followed by the standard enzyme assay as described earlier. The activity assayed in the absence of the enzyme inhibitors was defined as the control.
Determination of Kinetics Parameters
To obtain K m and V max of the phytase for sodium phytate, 0.5, 1.0, 1.5, 2.0, 3.0, and 4.0 mM of sodium phytate was mixed with 0.2 ml of the purified phytase (the final phytase concentration was 0.12 U/ml), respectively, followed by the standard enzyme assay as described above. K m, V max, and K cat values were obtained from a Lineweaver–Burk plot and expressed as the mean of the three different experiments.
Results and Discussion
Purification of Phytase
The extracellular phytase was purified from the supernatant prepared from the cell culture by ammonium sulfate fractionation, gel filtration chromatography (Sephadex™ G-75), and DEAE Sepharose Fast Flow anion-exchange chromatography. The elution profile of gel filtration chromatography indicates that peak 5 with the specific phytase activity from fractions 1 to 10 showed a single peak (Figure 1A) while the elution profile of DEAE Sepharose Fast Flow anion-exchange chromatography shows that peak 4 with the specific phytase activity from fractions 6 to 15 displayed one single sharp peak (Figure 1B). Therefore, the fractions were collected and concentrated by ultrafiltration. The results in Table 1 show that the enzyme was purified to homogeneity with a 7.2-fold increase in specific phytase activity with a yield of about 10.4% as compared to that in the supernatant.

Elution profiles of phytase from Kodamaea ohmeri BG3 on Sephadex™G-75 (A) and DEAE Sepharose fast flow anion exchange (B). (▪), Absorbance at 280 nm; (▴), specific activity.
Gel Electrophoresis
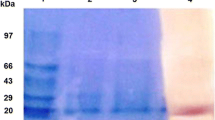
SDS-PAGE was used to determine protein purity and estimate molecular mass of the finally concentrated elute as described by Laemmli (1970). The results in Figure 2 indicate that there was one single protein band from the final concentrated elute and the relative molecular mass of the purified phytase was estimated to be 98.2 kDa by SDS-PAGE. It has been reported that the majority of yeast extracellular phytase had a molecular mass greater than 95 kDa; even the phytase produced by S. castellii had a molecular mass of 490 kDa with a glycosylation rate of approximately 31%. The structure of the deglycosylated protein was tetrameric, with one large subunit (MW 125,000) and three identical small subunits (MW 70,000) (Segueilha et al. 1992; Pandey et al. 2001).

SDS-PAGE (12%) of the fractions showing phytase activity obtained during the purification. Lane M, marker proteins with relative molecular masses indicated on the right; lane 1, eluate from DEAE Sepharose fast flow anion exchange chromatography; lane 2, crude enzyme.
Estimation of Molecular Mass of the Purified Phytase by Gel Filtration Chromatography
A calibration curve (molecular mass vs. V e/V o) was made according to V o and V e values and molecular mass of the individual protein standards as described in Materials and Methods. Molecular mass of the purified phytase was estimated to be 92.9 kDa via gel filtration chromatography according to the curve, as the elution volume of the purified phytase was 125 ml (data not shown). The results also indicate that the phytase from the marine yeast strain was a 92.8 kDa protein monomer, which was similar to the results of SDS-PAGE.
Cloning of Full Length Sequence of the Gene Encoding Phytase in Kodamaea ohmeri BG3
After sequencing of the cloned partial DNA fragment (accession number EU009483) as described in Materials and Methods, the full-length sequence of the gene encoding phytase in Kodamaea ohmeri BG3 was cloned via the SMART™ RACE cDNA Amplification Kit (Clontech). From the full length DNA sequence (EU 082006), amino acid sequence was deduced by using software offered in NCBI. Multiple alignments of the amino acid sequence were produced via DNAMAN 6.0 based on amino acid sequences of phytases from D. castellii (ABN04184), A. adeninivorans (AM231307) (Kaur et al. 2007), P. guilliermondii (CAL69849), and the amino acid sequence deduced from the cloned full-length DNA sequence (Figure 3). The results in Figure 3 show that amino acid sequence deduced from the cloned full-length DNA sequence also contained the N-terminal and C-terminal motifs RHGAR X PT and HD, which are characteristic for histidine acid phosphatases. This means that the cloned full-length DNA sequence indeed encoded histidine acid phosphatases. As shown below, the purified phytase from Kodamaea ohmeri BG3 was also inhibited by phenylgloxal hydrate, a specific inhibitor of histidine acid phosphatases.

Multiple alignment of protein sequences encoding yeast phytases. The phytases are referenced by their databases. Multiple sequence alignment of proteins was carried out using the DNAMAN 6.0 method based on amino acid sequences of phytases from Debaryomyces castellii (ABN04184), Arxula adeninivorans (AM231307) (Kaur et al., 2007), and Pichia guilliermondii (CAL69849) and the amino acid sequence deduced from the cloned full-length DNA fragment of the yeast strain used in this study. Identical residues are indicated by black boxes. The N-terminal and C-terminal motifs RHGAR X PT and HD, characteristic for histidine acid phosphatases, are also marked.
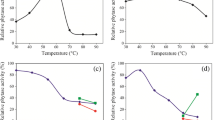
Optimum Temperature and Thermal Stability
The phytase activity measured as a function of temperature from 30°C to 80°C shows that the activity was highest at 65°C (Figure 4). Thermostability is considered an important and useful criterion for industrial application of phytase. For example, thermostability is a prerequisite for the successful application of enzymes in marine animal feeds that are exposed to 60°C to 90°C during the pelleting process. Therefore, thermostability was investigated by preincubating the enzyme in the same buffer as described in Materials and Methods for 1 h and the remaining activity was determined. As shown in Figure 4, the residual phytase activity still maintained 96.3% of the control after treatment at 60°C for 1 h, indicating that the enzyme was stable up to 60°C. Figure 4 also reveals that the enzyme was inactivated rapidly at temperatures higher than 60°C and was almost inactivated at 70°C within 1 h. From these results, the phytase seemed to have considerable thermostability. Phytase, in general, shows high activity in the temperature range of 50°C to 70°C while the optimum temperature is mostly between 45°C and 60°C (Vats and Banerjee 2004). For example, the phytase produced by by S. castellii exhibited an uncommon preference for high temperatures, with optimum activity at 77°C and thermostability up to 74°C (Segueilha et al. 1992; Pandey et al. 2001). This means that the thermostability of phytase from the marine yeast was lower than that from S. castellii.

Effects of different temperature on and activity (♦) and stability (▴) of the phytase. Temperature stability of the purified enzyme was tested by preincubating the enzyme at different temperatures (20, 40, 60, 65, and 70°C) for 1 h; the residual activity was measured immediately as described in the text. Here, preincubated sample at 0°C was used a reference to calculate the residual activity. Data are given as means ± SD, n = 3.
Optimum pH and pH Stability
Phytase activity was measured at various pH values in buffers with the same ionic concentrations. Our results (Figure 5) show that the maximum activity was observed at pH 5.0. pH stability was tested via 6-h preincubation of the purified enzyme in appropriate buffers that had the same ionic concentrations at different pH values ranging from 4.0 to 10.0 at 0°C. The remaining activities of phytase were measured immediately after this treatment with the standard method as mentioned earlier. It can be seen from the results in Figure 5 that the activity profile of the enzyme was stable from pH 3.0 to pH 9.0. For example, greater than 94.0% the residual activity was maintained after treatment at pH from 3.0 to 9.0 and 0°C for 6 h. These results suggest that the enzyme was very stable in the pH range of 3.0 to 9.0. Generally, the phytases from the bacterial source have optimum pH in the neutral to alkaline range, while in fungi the optimum pH range is 2.5 to 6.0 and the stability of phytase decreased dramatically above pH 7.5 and below pH 3.0, with a few exceptions of lower pH optima of 2.0 (Han et al. 1999; Pandey et al. 2001; Vats and Banerjee 2004). For example, the optimum pH of phytase produced by S. castellii was 4.4 (Segueilha et al. 1992). However, to date little has been known regarding pH stability of phytase secreted by terrestrial yeasts. The results indicate that the phytase from the marine yeast had greater pH stability in the alkaline range than that from other fungi, which may be related to the marine environment where the yeast strain was isolated.

Effects of different pH on activity (♦) and stability (▴) of the phytase. The pH stability was tested via 6 h preincubation of the purified enzyme in appropriate buffers that had the same ionic concentrations at different pH values ranging from 3.0 to 10.0 at 4°C. The remaining activities of phytase were measured immediately after this treatment with the standard method as described in the text. The phytase activity of the finally concentrated elute without preincubation was regarded as 100%. Data are given as means ± SD, n = 3.
Effects of Different Cations and Enzyme Inhibitors on Activity of the Purified Phytase
Mn2+, Ca2+, K+, Li+, Na+, Ba2+ Mg2+ and Co2+ (at concentrations of 5.0 mM) stimulated the activity of the purified phytase. However, Zn2+, Hg2+, Ag+, Cu2+, Fe3+ and Fe2+ (at a concentrations of 5.0 mM) acted as inhibitors in decreasing activity of the purified phytase, with Hg2+ (at a concentration of 5.0 mM) showing the lowest level (0.8%) (Table 2). However, the phytase from A. adeninivorans was only slightly sensitive to low concentrations of various ions (Mg2+, Ca2+, and Zn2+) (Sano et al. 1999). However, binding of two calcium ions to high-affinity calcium binding sites of bacterial phytase resulted in a dramatic increase in thermostability by joining loop segments remote in the amino acid sequence. The phytase from Klebsiella oxytoca MO-3 was strongly inhibited by NaF, Zn2+; Fe2+, and Cu2+ (Jareonkitmongkol et al. 1997) while the phytase from one strain of A. niger was inhibited by Cu2+, Zn2+, Hg2+, Sn2+, and Cd2+ ions and activated by Ca2+, Mg2+, and Mn2+ ions (Dvorakova et al. 1997).
Table 3 depicts the effects observed in the presence of enzyme inhibitors of the purified phytase. The presence of the chelating agents EDTA and 1,10-phenanthroline did not affect enzyme activity, demonstrating that the purified enzyme was not a metalloenzyme (Ramirez-Zavala et al. 2004). The enzyme activity was slightly inhibited by PMSF and iodoacetic acid and was strongly inhibited by phenylgloxal hydrate, which is a specific inhibitor of histidine acid phosphatases. The phytase from Klebsiella oxytoca MO-3 was also not inhibited by EDTA or N-ethylmaleimide (Jareonkitmongkol et al. 1997). It has been reported that a purified phytase from B. subtilis strain VTT E-68013 was readily inhibited by EDTA (Kerovuo et al. 1998). However, to date little is known about the effects of enzyme inhibitors on phytase activity produced by terrestrial yeasts.
Kinetics Parameters
Lineweaver–Burk plots in this study show that apparent K m and V max values of the enzyme for sodium phytate were 1.45 mM and 0.083 μmol/ml · min (data not shown), respectively. The results also show that K cat value of the purified phytase was 0.93 s-1 (data not shown).
References
Adams A, Gottschling DE, Kaiser CA, Stearns T (1998) Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. (Cold Spring Habbor, NY: Cold Spring Harbor Laboratory Press) pp 84–88
Bindu S, Somashekar D, Joseph R (1998) A comparative study on permeabilization treatments for in situ determination of phytase of Rhodotorula gracilis. Lett Appl Microbiol 27, 336–340
Bradford MM (1976) A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248–253
Chi Z, Kohlwein SD, Paltauf F (1999) Role of phosphatidylinositol (PI) in ethanol production and ethanol tolerance by highly ethanol producing yeasts. J Indus Microbiol Biotechnol 22, 58–63
Chi Z, Liu Z, Gao L, Gong F, Ma C, Wang X, Li H (2006) Marine yeasts and their applications in mariculture. J Ocean Univ China 5, 251–256
Dvorakova J, Volfova O, Kopecky J (1997) Characterization of phytase produced by Aspergillus niger. Folia Microbiol 42, 349–352
George V, Diwan AM (1983) Simultaneous staining of proteins during polyacrylamide gel electrophoresis in acidic gels by countermigration of Coomassie brilliant blue R-250. Anal Biochem 132, 481–483
Haefner S, Knietsch A, Scholten E, Braun J, Lohscheidt M, Zelder O (2005) Biotechnological production and applications of phytases. Appl Microbiol Biotechnol 68, 588–597
Han YM, Lei XG (1999) Role of glycosylation in the functional expression of an Aspergillus niger phytase (phyA) in Pichia pastoris. Arch Biochem Biophys 364, 83–90
Han YM, Wilson DB, Lei XG (1999) Expression of an Aspergillus niger phytase gene (phyA) in Saccharomyces cerevisiae. Appl Environ Microbiol 65, 1915–1918
Hirimuthugoda NY, Chi Z, Li X, Wang L, Wu L (2006) Diversity of phytase-producing marine yeasts. Ciencias Marinas 32, 673–682
Jareonkitmongkol S, Ohya M, Watanab R, Takagi H, Nakamori S (1997) Partial purification of phytase from a soil isolate bacterium, Klebsiella oxytoca MO-3. J Ferment Bioeng 83, 393–394
Kaur P, Lingner A, Singh B, Boer E, Polajeva J, Steinborn G, Bode R, Gellissen G, Satyanarayana T, Kunze G (2007) APHO1 from the yeast Arxula adeninivorans encodes an acid phosphatase of broad substrate specificity. J Antonie Van Leeuwenhoek 91, 45–55
Kerovuo J, Lauraeus M, Nurminen P, Kalkkinen N, Apajalahti J (1998) Isolation, characterization, molecular gene cloning, and sequencing of a novel phytase from Bacillus subtilis. Appl Environ Microbiol 64, 2079–2085
Laemmli UK (1970) Cleavage of structural proteins during assembly of head of bacteriophage T4. Nature 227, 680–685
Mayer AF, Hellmuth K, Schlieker H, Lopez-Ulibarri R, Oertel S, Dahlems U, Strasser AWM, Van-Loon APGM (1999) An expression system matures: a highly efficient and cost-effective process for phytase production by recombinant strains of Hansenula polymorpha. Biotechnol Bioeng 63, 373–381
Nakamura T, Suzuki T, Tokuda J, Kato N, Sakai Y, Mochizuki D, Takhashi H (1999) Secretory manufacture of Schwanniomyces occidentalis phytase using a Candida boidinii host. Eur Patent Appl Ep 931, 837 July 28
Pandey A, Szakacs G, Soccol CR, Rodriguez-Leond JA, Soccol VT (2001) Production, purification and properties of microbial phytases. Bioresource Technol 77, 203–214
Ramirez-Zavala B, Mercado-Flores Y, Hernadez-Rodriguez C, Villa-Tanaca L (2004) Purification and characterization of lysine aminopeptidase from Kluyveromyces marxiamus. FEMS Microbiol Lett 235, 369–375
Sano K, Fukuhara H, Nakamura Y (1999) Phytase of the yeast Arxula adeninivorans. Biotechnol Lett 21, 33–38
Segueilha L, Lambrechts C, Boze H, Moulin G, Galzy P (1992) Purification and properties of the phytase from Schwanniomycescastellii. J Ferment Bioeng 74, 7–11
Vats P, Banerjee UC (2004) Production studies and catalytic properties of phytases (myo-inositolhexakisphosphate phosphohydrolases): an overview. Enzyme Microb Technol 35, 3–14
Vohara A, Satyanarayana T (2004) A cost effective molasses medium for enhanced cell bound phytase production by Pichia anomala. J Appl Microbiol 97, 471–476
Acknowledgments
This research was supported by Hi-Tech Research and Development Program of China (863), the grant No is 2006AA09Z403.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, X., Chi, Z., Liu, Z. et al. Purification and Characterization of Extracellular Phytase from a Marine Yeast Kodamaea ohmeri BG3. Mar Biotechnol 10, 190–197 (2008). https://doi.org/10.1007/s10126-007-9051-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-007-9051-z