
Abstract
Polyurethane foam (PUF) supplemented with various agar media was used in situ to trap marine bacteria and it consequently provided a substrate on which they could be cultivated while exposed to natural seawater in the coral reef area. The bacterial population on the PUF blocks was analyzed by denaturing gradient gel electrophoresis (DGGE) of polymerase chain reaction (PCR)-amplified 16S rDNA fragments. Changing the composition of the cultivation medium in the PUF blocks and selecting different sampling sites resulted in different bacteria being detected on the PUF blocks. For example, iron-utilizing (IU) bacteria, siderophore-producing (SP) bacteria, and petroleum-degrading (PD) bacteria were isolated from PUF blocks and it was discovered that IU and SP contained iron and PD contained hydrocarbon. This method opens up the possibility for isolating novel and useful marine bacteria.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
More than 99% of naturally occurring microorganisms remain uncultured (Hugenholtz et al., 1998), because methods for isolation of microorganisms from natural environments still remain to be established. Previous work reported that carriers such as glass submerged in the sea become populated by periphytic bacteria (Corpe, 1973). However, there are few reports that the carrier soaked with agar media can be used as a tool for the isolation of marine bacteria.
This article describes a potentially useful enrichment culture method using polyurethane foam soaked with various culture media (PUF-media). This method may provide a three-dimensional culture environment that is composed of various different subenvironments for microorganisms. The subenvironments have different conditions for growth of microorganisms, such as pH, oxygen concentration, and so forth. PUF has been used as an effective microbe carrier in a down-flow microfilm bioreactor (Araki et al., 1999).
Enrichment culture is a method for isolating bacteria from the environment. We suggest that it may be possible to stimulate the growth of microorganisms possessing such expected functions as hydrocarbon degradation or metal utilization by supplementing the cultivation medium with hydrocarbons or some metals. This method is good for isolating novel and useful marine bacteria.
Materials and Methods
On-Site PUF Culture
PUF blocks (5 × 5 × 7 cm) were autoclaved in a glass beaker, soaked in 100 ml of a hot agar medium, pressed several times with a sterile spoon so that the medium could penetrate the center of each block, and then they were cooled and stored in a sterile package. The composition of each agar medium used was as follows: (1) MA, marine broth (Difco) containing 1.5% agar; (2) MA + Ca, marine broth (Difco) with 1% CaCO3 containing 1.5% agar; (3) A + Hex (n-hexadecane), 30.0 g of NaCl, 0.5 g of KH2PO4, 1.0 g of K2HPO4, 0.5 g of MgSO4·7H2O, 0.2 g of CaCl2·2H2O, 0.3 g of KCl, 0.01 g of FeCl2·nH2O, 1000 ml of distilled water, 1.0 g of l-alanine, 15.0 g of agar, and 1 ml of n-hexadecane at pH 7.5; (4) NSW + Kero (kerosene), 1.0 g of NH4NO3, 0.02 g of ferric citrate, 0.02 g of K2HPO4, 0.5 g of yeast extract, 800 ml of filtered seawater, 200 ml of distilled water, 15.0 g of agar, and 5 ml of kerosene at pH 7.8; and (5) MA + Fe, marine broth (Difco) with 0.3% iron (III) citrate hydrate containing 1.5% agar. Enrichment cultures using A + Hex (n-hexadecane) and NSW + Kero (kerosene) liquid media have been reported as a method for isolating hydrocarbon-degrading bacteria (Fujisawa et al., 1977; Higashihara et al., 1978).
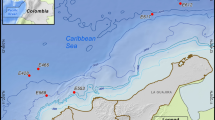
PUF blocks loaded with agar media were tied to arope at intervals of 80 cm with one PUF block unsupplemented with agar medium as a blank control. This rope with the series of PUF blocks was positioned about 5 to 6 m below the surface of the sea at Pohnpei in Micronesia for 3 days from December 4, 2001. The sampling points over the coral reefs were as follows: point 1, the channel near the reef edge (6°58′75″ N, 158°07′60″ E); point 2, near the mangroves (6°59′30″ N, 158°11′ E); and point 3, around the patch reef between 1 and 2 (6°58′85″ N, 158°10′ 02″ E). Three days after the deployment, PUF blocks were recovered, immersed in TE buffer (10 mM Tris-HCl and 1 mM EDTA at pH 8.0) and stored at ambient temperature for 4 days until their return to the laboratory with subsequent storage at –20°C.
Scanning Electron Microscopy (SEM)
After in situ incubation, PUF cubes were fixed in 2.5% glutaraldehyde solution (in seawater) for 24 h. The samples were dehydrated using ethanol series after post-fixation using osmium tetroxide. Dehydrated samples were critical-point dried (CO2), mounted, and sputter-coated with platinum-palladium. SEM observation was performed on a Hitachi S-2500 electron microscope at 15 kV.
DNA Extraction
Each PUF block (1 cm3) was ground with a mortar and pestle in the TE buffer (1 ml) and transferred to a 2-ml sample tube. DNA extraction was carried out by using TE-equilibrated phenol (pH 7.5 to 8.0), phenol–chloroform–isoamyl alcohol (25:24:1, by vol), and chloroform–isoamyl alcohol (24:1, vol/vol). The DNA was precipitated with 2-propanol, before the extracted DNA was purified using a GFX genomic blood purification kit (Amersham Pharmacia Biotech).
Polymerase Chain Reaction (PCR) for Denaturing Gradient Gel Electrophoresis (DGGE) Analysis
The PCR-DGGE method was used to analyze the bacterial population of each PUF block, with a PCR primer set and touchdown PCR protocol similar to those used by Muyzer et al. (1993, 1996). Variable region 3 of 16S rDNA from the bacterial community in each PUF block was amplified by touchdown PCR with two primers. These primers for PCR were 341F, (5′-CCTACGGGAGGCAG CAG-3′), of which the 5′ end was attached to a GC-rich sequence (5′-CGCCCGCCGCGCGCGGC GGGCGGGGCGGGGGCACGGGGGG-3′) and 534R (5′-ATTACCGCGGCTGCTGG-3′). They respectively corresponded to 341–357 and 517–534 of Escherichia coli 16S rDNA sequences. The reaction mixtures used for PCR contained (per 50 μl) 5 μl of a 10× PCR buffer containing MgCl2, 4 μl of a dNTP solution, the primers (341F-GC and 534R) at a concentration of 25 μM, 0.25 U of Ampli Taq Gold (Applied Biosystems), and 10 ng of DNA. The touchdown PCR protocol was as follows: The initial denaturation stage was started at 94°C for9 min, followed by 18 cycles at 94°C for 1 min, 64°C for 1 min (this annealing temperature was decreased by 1°C every two cycles), and then at 72°C for 2 min, followed by 30 cycles at 94°C for1min, 55°C for 1 min, and 72°C for 2 min, before the final extension was carried out at 72°C for 10 min. After the amplified PCR products were checked using 3% agarose gel electrophoresis, DGGE was carried out with the D-code system (Bio-Rad Laboratories), using a TAE buffer and 10% acrylamide gel [a 40% acrylamide stock solution (acrylamide–bis-acrylamide of 37.5:1)] with a 30% to 55% denaturing gradient of urea and formamideat 60°C for 3.5 h at a constant voltage of 200V. After electrophoresis, the gel was soaked in SYBRTM Green I nucleic acid gel stain (1:10,000 dilution; Molecular Probes, Inc.) for 30 min and then photographed on a UV transilluminator with a CCD camera. Selected DGGE bands were excised from the gel with a surgical blade and transferredinto fresh sterile microtubes. DNA was extracted from the excised gel with TE buffer. The extracted DNA was purified, and DGGE was repeated until there was only a single band without any trace from other bands. The partial sequence of the DNA from this band was analyzed by an ABI 3700 automated sequencer(Applied Biosystems), using the ABI Prism(R) BigDye™ primer cycle sequencing kit (Applied Biosystems). The closest relative to each sequence was obtained by a BLAST (Altschul etal., 1997) in DNA database (DDBJ/EMBL/Genbank). Multiple alignment and construction of phylogenetic trees by the neighbor-joining method (Saitou and Nei, 1987) were performed with the CLUSTAL W computer program (Thompson et al., 1994).
Isolation of Microorganisms
Each PUF was cut into small pieces (1 cm3) and homogenized with 5ml of sterile seawater using a glass rod. The homogenate was diluted 1:100 with sterile seawater. Fifty microliters of the diluted homogenate was spread onto four types of agar medium: MA + CA; A + Hex; NSW + Kero; and 1/10MA: 3.74 g of marine broth, 750 ml of filtered seawater, 250 ml of distilled water, and 15.0 g of agar.
Identification of Isolated Bacteria
Genomic DNA of the isolated strains was extracted by using a Puregene DNA extraction kit (Gentra Systems). PCR amplification of the 16S rDNA was performed by using forward primer 341F and reverse primer 907R (5′-CCGTCAATTCATTTGAGTTT-3′) under the same conditions as those used for touchdown PCR. After purifying the PCR product with a QIAquick PCR purification kit (QIAGEN), the purified PCR products were sequenced and identified via the same procedure as that described for DGGE.
Detection of Siderophores
The chrome azurol S (CAS) assay was used to detect siderophores (Schwyn and Neilands, 1987). On CAS agar plates, siderophore-producing bacteria formed colonies with an orange halo.
Competitive PCR
The competitor DNA that has a sequence of two bacterial primers (9F and 534R) each attaching to one end was constructed by using a competitive DNA construction kit (Takara Bio) according to the manufacturer's manual. Bacteria specific primer, 9F and universal primer, 534R were used to amplify bacterial 16S rDNA. One nanogram of extracted DNA from each sample and known copies of the competitor DNA were added into the same PCR tube and amplified with AmpliTaq Gold (Applied Biosystems) under the following conditions: denaturation at 94°C for 9 min, and then 40 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 2 min, and a final extension step of 72°C for 10 min. The amplified PCR products were stained with ethidium bromide and checked using 3% agarose gel electrophoresis. The gel image was taken with an FMBIO II Multi-View fluorescent image analyzer (Hitachi Software Engineering Co.), and the fluorescence intensity of the amplified DNA band was analyzed by FMBIO Analysis Version 6.0 software (Hitachi Software Engineering Co.). Based on the obtained fluorescence intensity, the copy number of 16S rDNA in each sample was calculated via the following equation:
where C is the copy number of the DNA competitor, T is the copy number of the objective (template) DNA, a is the gradient of the plotted line, and log C 0 is the y-intercept.
Results and Discussion
On-Site PUF Block Culture
A preliminary examination of on-site PUF block culture was carried out in Okinawa. The agar that had been loaded into the PUF blocks was almost completely retained after 3 days of the on-site culture, apparently protected from environmental bacterial degradation. The PUF blocks cultured on site with the agar medium displayed distinct colors (Figure 1) and distinct smells when compared to the PUF blocks without the medium. It is assumed that agar filled the internal pores of the PUF of the PUF blocks, enabling environmental microbes to attach to the agar surface and to grow on the components in the medium. The center of each PUF block seemed to be anoxic, indicated by black coloration and the presence of anaerobic bacteria, which was suggested by 16S rDNA gene sequencing that showed strains related to known anaerobes. Growth of the bacteria on the PUF was examined by SEM (Figure 2).

PUF samples loaded with the different media displayed distinct differences in color and smell when compared with the control samples (sampling point 2).

SEM image of PUF-MA + Ca. The PUF cube was obtained in Okinawa, 2002. (a) Scale bar = 150 mm. (b) Scale bar = 7.5 mm. This is the surface of the cross section.
Population Analysis by the PCR-DGGE Method
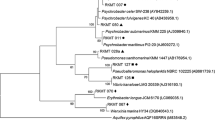
Partial 16S rDNA fragments were separated by DGGE, and 67 bands were sequenced (Figure 3). Table 1 lists the bacterial species that exhibited the highest nucleotide identity to each band from DGGE. Two sequences, PO-11(o)-7 (sampling point 2, MA + Fe medium) and PO-7-7 (sampling point 2, MA + Ca medium), were identified as close relatives (although only 87% and 95% similarity, respectively) of the uncultured bacterium [AY038542] which had previously been identified as a likely candidate division. The DGGE band, PO-13-1 (sampling point 3, MA + Ca medium) was closely related to the TM7 candidate division [AF269024] (Hugenholtz et al., 2001).

DGGE analysis of bacterial 16S rDNA fragments from the PUF samples at Pohnpei. Lanes: 1–5, sampling point 1; 6–11, sampling point 2; 12–16, sampling point 3; 1, 6, and 12, MA; 2, 7, and 13, MA + Ca; 3, 8, and 14, control; 4, 9, and 15, A + Hex; 5, 10, and 16, NSW + Kero; 11o, MA + Fe (outside of PUF); 11i, MA + Fe (inside of PUF).
We selected CaCO3 as substance for PUF media because Ca is required for the bones of various marine organisms in different habitants in the coral reef area. When MA + Ca was applied as media, the microbial diversity increased. Alphaproteobacteria, Deltaproteobacteria, Epsilonproteobacteria, Gammaproteobacteria, Bacteroidetes, Firmicutes, TM7, and unclassified bacteria were detected. We plan to investigate the relationships between the habitants and the bacteria that concentrate in the habitats in the near future.
Vibrio spp. were predominant at both sampling points 1 and 2, but could not be detected at sampling point 3. Alphaproteobacteria, Epsilonproteobacteria, Gammaproteobacteria, and Bacteroidetes were each detected as DGGE bands at all three sampling points.
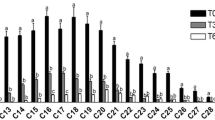
The difference in division level of the bacterial community associated with the PUF samples by sampling point is shown in Figure 4. Firmicutes were predominant at sampling points 2 and 3, but could not be detected at sampling point 1 by DGGE. The Firmicutes detected as DGGE bands were almost all (13 out of 14) related to Fusibacter paucivorans [AF0 50099] which had been reported to be an anaerobic thiosulfate-reducing bacterium (Ravot et al., 1999).

Histogram illustrating the division-level diversity of the partial 16S rDNA bacterial sequence of DGGE bands associated with the PUF samples from the three sampling points. Sampling point 1, the channel near the reef edge; sampling point 2, near the mangroves; sampling point 3, around the patch reef between 1 and 2.
Isolated Bacteria
One hundred and twenty-two isolates were obtained, and were identified via 16S rDNA gene sequence analysis.
All the bacteria (19 strains, 14 of which sequenced) isolated from the PUF samples soaked with the MA + Fe medium were found to produce siderophores determined by CAS assay (Schwyn and Neilands,1987), whereas 6 out of the 14 strains (12 of which were sequenced) of bacteria isolated from the PUF soaked in the MA medium were found to be CAS-positive. Siderophores are low-molecular-mass, iron-chelating compounds that have medical and agricultural applications. Guan et al. (2001) have also reported on the isolation of siderophore-producing bacteria from the marine environment. Bacterial strain PO-47 [AB235413] was isolated from PUF MA+ Fe block. The PO-47 was identified as being closelyrelated (99% similarity) to Shewanella alga [AF006669], which has been reported to grow anaerobically, by dissimilatory Fe(III) reduction (Bowman et al., 1997). The PO-47 strain produced the cyclic dihydroxamate siderophore (about 1 g of bisucaberin from 4 liters of culture broth) (Yasumoto-Hirose etal., unpublished data). Bisucaberin was previously isolated from a marine bacterium, Alteromonas haloplanktis (Kameyama et al., 1987; Takahashi etal., 1987).
n-Alkane-degrading bacteria belonging to the Alcanivorax group were isolated from the PUF soaked in A + Hex (at sampling points 2 and 3) and in NSW + Kero (sampling point 3). The Alcanivorax genus has been reported to play an important role in the first step of crude oil biodegradation in a marine environment (Harayama et al., 1999). The Alcanivorax sp. was isolated from PUF containing hydrocarbon. These were isolated from various plates: the A+ Hex, the NSW + Kero, and the Ca plates. These results prompt speculation that bacteria grew on the PUF blocks by hydrocarbon degradation. It is possible that the bacteria were concentrated in the natural marine environment by the PUF. However, the bacteria on the PUF did not result simply from such concentration, but from enrichment by the media employed.
Ten sequences detected as DGGE bands were related to Epsilonproteobacteria. Bacterial strains PO-40 [AB235414] was identified as closely related (96% similarity) to an uncultured epsilon bacterium [AF235116], and also to DGGE bands PO-1-6 [AB235415].
One liter of seawater at each PUF sampling area was collected, filtered through a sterile 0.22-μm filter, DNA extracted, and DGGE analyzed, and the sample was compared with the PUF samples in the marine environment, showing different band patterns (Yasumoto-Hirose et al., unpublished data). This media-supplemented PUF method likely provides a three-dimensional culture environment that is composed of various different subenvironments for microorganisms. The microenvironment in the media-supplemented PUF has different conditions for growth of microorganisms, such as pH, oxygen concentration, and so forth. Many strains detected as DGGE bands were related to anaerobic bacteria, but isolation of the anaerobic bacteria remains to be done.
Competitive PCR
Competitive PCR was carried out for quantifying 16S rDNA to estimate the bacterial population on each PUF block. The DNA extracted from each PUF was used. The number of copies of 16S rDNA per 1 cm3 of PUF was calculated (Figure 5). Although the number of copies did not equal the number of bacteria, the difference in the numbers of copies on PUF blocks with or without a medium was observed. Although the same amount (1 ng) of extracted DNA was used as template DNA for the competitive PCR, the PUF blocks without a medium (control PUF blocks) showed fewer than 10 copies per 1 ng DNA by competitive PCR; that is, the number of copies of bacteria 16S rDNA in 1 cm3 PUF was less than 103 to 104. This low number of copies/cm3 might be a result of attachment of eukaryotes on the control PUF. On the other hand, the PUF blocks soaked with media contained approximately 105 to 109 copies per 1 cm3 of PUF. Among medium-soaked PUFs, a larger number of copies 16S rDNA (108 order) was detected in PUF-MA (Figure 5). PUF-A + Hex showed a smaller number among tested media. PUF-NSW + Kero showed a large number at points 1 and 2, but a small number at point 3. It was interesting that PUF-MA + Fe showed a larger number of copies (PUF-MA + Fe(i), 1.3 × 108 copies/1 cm3 of PUF; PUF-MA + Fe(o), 4.2 × 107 copies/1iter cm3 of PUF) than PUF-MA + Ca (5.8 × 106 copies/1 cm3 of PUF) at sampling point 2. The two kinds of PUFs used MA for basal medium. In addition, both the bacterial diversity detected on DGGE bands and the diversity of the isolated strains on PUF-MA + Ca were higher than the diversity of bacteria on PUF-MA + Fe (Tables 1 and 2). Different media containing various compounds have different selective pressures on bacterial growth on the PUF. The growth of hydrocarbon-degrading bacteria was likely enhanced on the PUF with hydrocarbons (hexadecane and kerosene) more than the growth of other heterotrophic bacteria.

The copy number of 16S rDNA of the microbial consortia attached to each medium-soaked PUF (1–16) was determined by competitive PCR. The control PUF samples without any medium (3, 8, and 14).
The methods and procedures reported here make it possible to collect naturally occurring marine bacteria relatively simply and economically. Even if the population of the bacteria in seawater is quite low, the PUF block is a powerful tool to enrich various types of bacteria with interesting properties in natural environments. After enrichment culture using PUF, the target bacteria can be isolated by conventional agar plate media containing certain selective substrate. In our study, we isolated hydrocarbon-degrading bacteria from PUF blocks containing hydrocarbon and siderophore-producing bacteria from those containing iron. We intend to study further whether the method can be applied to other varieties of bacteria in different natural environments.
References
SF Altschul TL Madden AA Schaffer J Zhang Z Zhang W Miller DJ Lipman (1997) ArticleTitleGapped BLAST and PSI-BLAST: a new generation of protein database search programs Nucleic Acids Res 25 3389–3402 Occurrence Handle10.1093/nar/25.17.3389
N Araki A Ohashi I Machdar H Harada (1999) ArticleTitleBehaviors of nitrifiers in a novel biofilm reactor employing hanging sponge cubes as attachment site Wat Sci Tech 39 23–31 Occurrence Handle10.1016/S0273-1223(99)00146-8
JP Bowman SA Mccammon DS Nichols JH Skerratt SM Rea PD Nichols TA McMeekin (1997) ArticleTitleShewanella gelidimarina sp. nov. and Shewanella frigimarina sp. nov., novel antarctic species with the ability to produce eicosapentaenoic acid (20:5ω3) and grow anaerobically by dissimilatory Fe (III) reduction Int J Syst Bacteriol 47 1040–1047
WA Crope (1973) Microfouling: the role of primary film forming marine bacteria RF Acker BF Brown JR Palma Particlede JR Iverson (Eds) Proceedings of the 3rd International Congress of Marine Corrosion and Fouling Northwestern University Press Evanston, IL 598–609
H Fujisawa M Murakami T Manabe (1977) ArticleTitleEcological studies on hydrocarbon-oxidizing bacteria in Japanese coastal waters-I Bull Jpn Soc Sci Fish 43 659–668
LL Guan K Kanoh K Kamino (2001) ArticleTitleEffect of exogenous siderophores on iron uptake activity of marine bacteria under iron-limited conditions Appl Environ Microbiol 67 1710–1717 Occurrence Handle10.1128/AEM.67.4.1710-1717.2001
S Harayama H Kishira Y Kasai K Shutsubo (1999) ArticleTitlePetroleum biodegradation in marine environments J Mol Microbiol Biotechnol 1 63–70
T Higashihara A Sato U Shimidu (1978) ArticleTitleAn MNP method for the enumeration of marine hydrocarbon degrading bacteria Bull Jpn Soc Sci Fish- 44 1127–1134
P Hugenholtz BM Goebel NR Pace (1998) ArticleTitleImpact of culture-independent studies on the emerging phylogenetic view of bacterial diversity J Bacteriol 180 4765–4774
P Hugenholtz GW Tyson RI Webb AM Wagner LL Blackall (2001) ArticleTitleInvestigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives Appl Environ Microbiol 67 411–419 Occurrence Handle10.1128/AEM.67.1.411-419.2001
T Kameyama A Takahashi S Kurasawa M Ishizuka Y Okami T Takeuchi H Umezawa (1987) ArticleTitleBisucaberin, a new siderophore, sensitizing tumor cells to macrophage-mediated cytolysis. I. Taxonomy of the producing organism, isolation and biological properties J Antibiot (Tokyo) 40 1664–1670
G Muyzer EC Waal Particlede AG Uitterlinden (1993) ArticleTitleProfiling of complex microbial populations by denaturing gradient gel electrophresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA Appl Environ Microbiol 59 695–700
G Muyzer S Hottenträger A Teske C Wawer (1996) Denaturing gradient gel electrophoresis of PCR-amplified 16S rDNA—a new molecular approach to analyse the genetic diversity of mixed communities ADL Akkermans JD Elsas Particlevan FJ Brujin Particlede (Eds) Molecular Microbial Ecology Manual, 3.4.4 Kluwer Academic Dordrecht 1–23
G Ravot M Magot ML Fardeau BKC Patel P Thomas JL Garcia B Ollivier (1999) ArticleTitleFusibacter paucivorans gen. nov., sp. nov., an anaerobic, thiosulfate-reducing bacterium from an oil-producing well Int J Syst Bacteriol 49 1141–1147 Occurrence Handle10.1099/00207713-49-3-1141
N Saitou M Nei (1987) ArticleTitleThe neighbor-joining method: a new method for reconstructing phylogenetic trees Mol Biol Evol 4 406–425
B Schwyn JB Neilands (1987) ArticleTitleUniversal chemical assay for the detection and determination of siderophores Anal Biochem 160 47–56 Occurrence Handle10.1016/0003-2697(87)90612-9
A Takahashi H Nakamura T Kameyama S Kurasawa H Naganawa Y Okami T Takeuchi H Umezawa Y Iitaka (1987) ArticleTitleBisucaberin, a new siderophore, sensitizing tumor cells to macrophage-mediated cytolysis. II. Physico-chemical properties and structure determination J Antibiot (Tokyo) 40 1671–1676
JD Thompson DG Higgins TJ Gibson (1994) ArticleTitleCLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice Nucleic Acids Res 22 4673–4680
Acknowledgments
We thank Izumi Yamashima for technical assistance. We also thank Minoru Yasumoto for sampling. This work was part of The Industrial Science and Technology Project for Technology Development of Biological Resources in Bioconsortia, which is supported by the New Energy and Industrial Technology Development Organization of Japan, as part of the project on Constructing the Genetic Resource Library of Unidentified Microbes Based on Genome Information which is supported by the Ministry of Economy, Trade and Industry of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yasumoto-Hirose, M., Nishijima, M., Ngirchechol, M.K. et al. Isolation of Marine Bacteria by In Situ Culture on Media-Supplemented Polyurethane Foam. Mar Biotechnol 8, 227–237 (2006). https://doi.org/10.1007/s10126-005-5015-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-005-5015-3