
Abstract
Karenia brevis (Davis) is the dinoflagellate responsible for nearly annual red tides in the Gulf of Mexico. Although the mechanisms regulating the growth and toxicity of this problematic organism are of considerable interest, little information is available on its molecular biology. We therefore constructed a complementary DNA library from which to gain insight into its expressed genome and to develop tools for studying its gene expression. Large-scale sequencing yielded 7001 high-quality expressed sequence tags (ESTs), which clustered into 5280 unique gene groups. The vast majority of genes expressed fell into a low-abundance class, with the highest expressed gene accounting for only 1% of the total ESTs. Approximately 29% of genes were found to have similarity to known sequences in other organisms after BLAST similarity comparisons to the GenBank public protein database using a cutoff of P < 10e−4. We identified for the first time in a dinoflagellate a suite of conserved eukaryotic genes involved in cell cycle control, intracellular signaling, and the transcription and translation machinery. At least 40% of gene clusters displayed single nucleotide polymorphisms, suggesting the presence of multiple gene copies. The average GC content of ESTs was 51%, with a slight preference for G or C in the third codon position (53.5%). The ESTs were used to develop an oligonucleotide microarray containing 4629 unique features and 3462 replicate probes. Microarray labeling has been optimized, and the microarray has been validated for probe specificity and reproducibility. This is the first information to be developed on the expressed genome of K. brevis and provides the basis from which to begin functional genomic studies on this harmful algal bloom species.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Dinoflagellates are unicellular protists most closely related to the ciliates and apicomplexa (Fast et al., 2002). Unique among eukaryotes, dinoflagellates have permanently condensed chromatin, but lack histones and nucleosomes typically involved in regulating chromosome condensation and gene expression. They also have evolved a unique mitosis in which the nuclear envelope remains intact and the mitotic spindle consists of extra nuclear microtubules that traverse the nucleus through cytoplasmic channels (Bhaud et al., 1999). Dinoflagellates typically possess large genomes (up to 200 pg/cell) that are generally considered to be haploid (Triplett et al., 1993; Santos and Coffroth, 2003).
Karenia brevis is a dinoflagellate whose expressed genome is of interest because of its role in producing the harmful algal blooms (HABs) or “red tides” that occur annually in the Gulf of Mexico. K. brevis blooms cause extensive fish kills, mortality among of protected marine mammals, and human illness through the production of highly potent neurotoxins known as brevetoxins. The K. brevis genome consists of 121 chromosomes (Walker, 1982) containing 100 pg of DNA per cell, or approximately 1 × 1011 bp (Kim and Martin, 1974; Rizzo, 1982; Sigee, 1986; Kamykowski et al., 1998). This is 30 times the size of the human genome; however, dinoflagellate chromosomes consist of a permanently condensed, genetically inactive central region with peripheral loops of B-DNA that protrude from this core and comprise the actively transcribed DNA (Sigee, 1984; Anderson et al., 1992; Bhaud et al., 1999). Therefore, although the size of the expressed genome of K. brevis is unknown, it is anticipated to be substantially smaller than its total genome size might predict.
Insight into the molecular mechanisms that control growth, toxicity, and persistence of K. brevis blooms is critical to understanding the formation of HABs; however, few investigations into the molecular biology of K. brevis exist. Antibody-based approaches have yielded some insight into K. brevis cell cycle control, with the identification of the central eukaryotic cell cycle regulator, cyclin-dependent kinase (Van Dolah and Leighfield, 1999), and its regulatory subunit cyclin (Barbier et al., 2003). Nonetheless the partners and cell cycle substrates of this central regulator remain unidentified, and the unique features of the dinoflagellate nucleus suggest that unusual mechanisms may have evolved. The basis for K. brevis toxicity is the production of brevetoxins, polyether toxins with structures that suggest synthesis through a polyketide synthase pathway. Yet investigations into polyketide synthase genes in K. brevis have resulted in ambiguity regarding the dinoflagellate-versus-bacterial contributions to polyketide synthase activity in this organism (Snyder et al., 2003). Antibody-based studies have also been used to identify stress proteins that may play a role in the adaptation and persistence of blooms under stressful environmental conditions (Miller-Morey and Van Dolah, 2004). However, the scope of such studies is limited by the availability of antibodies cross-reactive with dinoflagellate proteins. Thus the need is clear for genomic tools with which to study gene expression and regulation in this organism for which little molecular information is available.
Sequencing of complementary DNA libraries to generate expressed sequence tags (ESTs) is an effective means of discovering expressed genes in organisms for which genomic data are unavailable. ESTs serve as markers for genes expressed under specific conditions and can be used as probes in the recovery of full-length cDNA or genomic sequences, recognition of exon and intron boundaries, delineation of protein families, and development of probes for genomewide expression profiling. To this end we constructed a cDNA library to K. brevis and carried out large-scale sequencing to yield an expressed EST database containing 7001 ESTs and 5280 unique gene clusters. These ESTs were then used to develop an oligonucleotide microarray specific for K. brevis gene expression. Microarray technology provides the capacity to profile genomewide changes in gene expression in response to different exposure conditions, to identify genes involved in specific pathways on the basis of their coordinated responses, and to assign function to unknown genes on the basis of their induction in response to known challenges. This approach will greatly expand our understanding of K. brevis physiology at the molecular level and develop our understanding of the effects of modifications on the marine environment (Jenny et al., 2002). Here we present details of cDNA library construction, insight into the K. brevis genome as revealed by EST data analysis, and the development and validation of a DNA microarray for investigation of K. brevis functional genomics.
Materials and Methods
Strain and Culture Conditions of Cells
The Wilson isolate of K. brevis was used for this study. The growth and behavior of this isolate have been well studied during the approximately 50 years it has been in culture. Cells were maintained in batch culture in 1-L glass bottles with autoclaved, 20-μm-filtered seawater at 36 psu obtained from a seawater system at the Florida Institute of Technology field station at Vero Beach. Seawater was enriched with f/2 medium (Guillard, 1973). Cultures were maintained at 25° ± 1°C on a 16:8-hour light-dark cycle. Illumination from cool white lights was maintained at a photon flux density of 40 to 50 μE·m−2·s−1 (measured by Li-Cor 2π sensor).
Construction of cDNA Library
Cultures were harvested by centrifugation (1000g) during the logarithmic phase of growth at circadian time CT 16–18. This time was chosen to maximize the likelihood of expression of cell cycle genes, according to the known diel phasing of the K. brevis cell cycle (Van Dolah and Leighfield, 1999). Total RNA (2 mg) was isolated from approximately 20 L of K. brevis culture using Qiagen RNeasy columns. Then cDNAs were synthesized by oligo(dT) priming from poly(A) messenger RNA, size-selected (>400 bp), and directionally cloned into a λ Zap II vector system (Stratagene).
Clone Propagation, Plasmid Isolation, and EST Sequencing
Packaged phages were used to infect XL1-Blue cells, and mass in vivo excision of the pBluescript SK(−) phagemid from the λ ZAP II vector was performed with the ExAssist helper phage. Separate cultures of XL1-Blue MRF′ and SOLR cells were grown overnight in LB broth with supplements at 30°C. Cells were centrifuged (1000g) and resuspended in 10 mM MgSO4 to an OD600 of 1.0 (8 × 108 cells/ml). A portion of the λ bacteriophage library (3.9 × 107 pfu) was combined with XL1-Blue cells at a multiplicity of infection (MOI) of 1:10 λ phage–cell ratio. ExAssist helper phage (1 × 109 pfu) was added at a 10:1 helper phage–cell ratio to ensure that every cell was co-infected with λ phage and helper phage. Cells were incubated at 37°C to allow the phage to attach, transferred to LB broth, and incubated at 37°C with shaking. After 2.5 to 3 hours of incubation, cultures were heated (65°–70°C) for 20 minutes to lyse the cells.
The excised phagmids were transformed into SOLR cells, and individual colonies were grown on LB-ampicillin agar plates. The pBluescript DNA was purified with QIAprep Miniprep Kits using a QIAvac 96 Top Plate system or a BioRobot 9600 system (Qiagen). Sequencing reactions were carried out from the 5′ end of the cDNA insert using a universal T3 primer (5′-ATTAACCCTCACTAAAG-3′).
EST Database Analysis and Management
Lasergene SeqMan II software (DNAStar Inc.) was used to remove vector sequence, evaluate the quality of underlying sequencing trace data, and eliminate poor-quality ESTs using the following criteria: sequences shorter than 100 bp were eliminated, sequences with trace Phred threshold below 12 were removed, and sequences were terminated when more than 3Ns occurred in a 20-bp rolling window. Edited EST sequences were compared with the nonredundant GenBank sequence database using the basic local alignment search tool program (BLAST; Altschul et al., 1990) in its version for nucleotides (BLASTn) and amino acids (BLASTx). EST sequences were then clustered using the SeqMan II contig assembly process with a minimum cutoff value of 95% identity in a 50-bp window.
DNA Microarray Design and Validation
To facilitate design of 60-bp oligonucleotides that uniquely recognize gene sequences sharing identity as defined by BLASTx similarity scores, a second pass clustering was performed using identity criteria of 75% identity over 30 bp (Parcel Clustering Package Version 2.2.8). This contracted the number of unique contigs to 4629, to which 60-mer oligonucleotides were designed. The resulting oligonucleotide probes and controls were printed on glass slides using an Ink Jet–based printing method, yielding an 8455 feature array (Agilent Technologies), including 4629 primary probes to each of the unigenes, 3826 replicate probes to selected genes, and 192 hybridization controls. Two arrays were printed per 1 × 3-inch glass slide.
A self-versus-self hybridization was performed for determining probe specificity, array reproducibility, and microarray feature uniformity. Total RNA from K. brevis cultures was prepared using Tri-Reagent according to the manufacturer’s protocol. Following precipitation of the RNA, the pellet was resuspended and run through a Qiagen RNeasy column for removal of contaminating DNA and protein, and then total RNA was quantified by UV spectroscopy and qualified on an Agilent Bioanalyzer. Total RNA (350 ng) was amplified and labeled with the Cy3 and Cy5 dyes (PerkinElmer) with an Agilent low-input linear amplification kit according to manufacturer’s protocol. Following labeling we quantified amplified RNA by UV spectroscopy, and 350 ng each of Cy3 and Cy5 labeled targets were hybridized to the array for 17 hours at 60°C. After hybridization arrays were washed consecutively in solutions of 6× SSPE and 0.005% N-lauroylsarcosine and 0.06× SSPE and 0.005% N-laurel sarcosine for 1 minute each at room temperature. This was followed by a 30-second wash in a stabilization and drying solution (Agilent Technologies). Following the wash steps microarrays were imaged using an Agilent microarray scanner. The scan was extracted and normalized by a combination Linear/Lowess algorithm using Agilent Feature Extraction Version 7.5 software. Data were further evaluated for feature uniformity, signal intensity, and signal-background ratio, dye bias, and reproducibility of replicate probes using the Rosetta Luminator gene expression analysis system and Microsoft Excel.
Results and Discussion
Acquisition and Features of Generated ESTs
The K. brevis cDNA library contains 3.9 × 107 primary recombinants with an average insert size of 1.8 kb and an insert size range of 0.5 to 2.8 kb. A total of 9728 sequencing reactions were performed from the 5′ end of individual, randomly selected clones to obtain ESTs. After removal of sequences identified as low quality, 7001 sequences with a minimum of 100 bp of continuous sequence were retained for further analysis. The average size of ESTs in the database is approximately 700 bp. All EST sequences are publicly available on the joint NOAA/Medical University of South Carolina website (marinegenomics.org), along with their BLASTn and BLASTx search results, and have been submitted to the GenBank dbEST database (accession numbers CO59029–CO065717, CO517335–CO517390, CV173737–CV173976, and CV179548).
Assembly of ESTs into Unigene Clusters
Many difficulties exist with using raw EST sequence information to identify unique genes, as an EST represents only a partial sequence derived from a cDNA library clone corresponding to a single mRNA molecule that may be present in multiple copies. Artifacts from cDNA library construction and single pass sequencing reactions, long 3′ or 5′ untranslated regions, and the efficiency of reverse transcriptase often cause EST sequences to be relatively short, highly redundant, and error prone. Thus, to better assign gene identities, the ESTs were assembled into clusters with a minimum identity of 95% within a minimum 50-bp region of overlap. By this definition, contigs derived from the clustered sequences represent unique expressed genes. From the 7001 high-quality ESTs, 5280 unigene clusters were identified (Figure 1). Of these, 4399 contained single ESTs, representing 63% of the total ESTs analyzed. The remainder fell into 881 clusters with sizes ranging from 2 ESTs (576 clusters) to 31 ESTs (1 cluster). of these, 98% belonged to clusters containing 4 or fewer ESTs, indicating that the vast majority of genes expressed fell into a low-abundance class (Figure 2). The highest expressed sequence (whose cluster consisted of 31 ESTs) represents less than 1% of the total ESTs. Thus the overall redundancy of the library is low. This is reflected in the high rate of novel EST acquisition over the 9728 sequencing reactions performed (Figure 2). Similarly high sequence diversity is apparent in EST libraries generated to other dinoflagellate species, Lingulodinium polyedrum (1519 ESTs) and Amphidinium carterae (3380 ESTs; Bachvaroff et al., 2004). What fraction of the total expressed dinoflagellate genomes any of these EST collections represent is unknown at present. A total genome sequence has been completed for the apicomplexan Plasmodium (5300 genes; Gardner et al., 2002) and is underway for the ciliate Tetrahymena (25,000–30,000 genes expected; J. Carlton, TIGR, personal communication); thus the genome sizes of nearest relatives offer little insight into the sizes expected in dinoflagellates.

Cluster analysis of 7001 K. brevis ESTs. Unigene clusters are sorted as a function of cluster size (ESTs per cluster). Of a total of 5280 individual clusters, 4399 contain single ESTs, while 881 contain 2 to 31 ESTs.

Redundancy of the K. brevis ESTs. Number of unique sequences plotted against the number of total sequences.
Overview of Genes Identified by Database Search
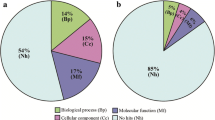
The 5280 contigs were compared with the public nonredundant protein sequence database using the BLASTx search algorithm. Using a cutoff P value of less than 10e−4, 1556 (29%) showed similarity to previously identified genes from a wide variety of organisms. Of these, the largest number are involved in metabolism (23%), signal transduction (20%), transcription/translation (15%), and structure/cytoskeleton (11%) (Figure 3).

Functional classification of 1556 EST clusters that showed similarity to previously identified genes. Distribution of functional classes excludes clusters of no known similarity.
Only 24 ESTs were present at 10 or more copies (Figure 4). The highest expressed gene in the library was that for fucoxanthin chlorophyll a/c binding protein, with 50 copies present (0.7% of the total ESTs). K. brevis is the only fucoxanthin-containing dinoflagellate for which gene expression data are available. However, among the peridinin-containing dinoflagellates for which data are available, the functionally analogous peridinin chlorophyll binding protein is also among the most highly expressed genes (Bachvaroff et al., 2004). Interestingly, flavodoxin, involved in the light reactions of photosynthesis, was the only other highly expressed photosytem gene identified in K. brevis (17 copies). Also among the highest expressers were s-adenosyl methionine synthase, adenosylhomocysteinase, and glutamine synthase, all components of amino acid biosynthetic pathways. These genes were also found to be among the highest expressed sequences in Lingulodinium (Bachvaroff et al., 2004). Cytoskeletal proteins were also represented in the highly expressed group, with actin present in 15 copies and α-tubulin in 34 copies. Its partner β-tubulin was found in only 3 copies, yet both α-tubulin and β-tubulin have been identified in the cytoskeleton of K. brevis by immunofluorescence and localization studies, suggesting the normal α, β dimer is present (Barbier, M., Miller, J., Morton, S.L., and Van Dolah, F.M., manuscript submitted).

Twenty-four most abundant genes.
Cell Cycle Genes
Despite their importance in understanding mechanisms regulating proliferation, little molecular information is available on dinoflagellate cell cycle genes. The K. brevis EST database provides the first collection of cell cycle genes in a dinoflagellate (Table 1). Cell cycle genes identified include the central cell cycle regulator cyclin-dependent kinase (CDC2). Its partner, cyclin, was not found, despite antibody-based evidence for its existence in dinoflagellates (Barbier et al., 2003; Wong et al., 1997); however, this is not surprising as even among mammals cyclin sequences are not highly conserved and cyclins were generally identified by complementation in yeast (Lew et al., 1991). Overall, S-phase-specific genes involved in the DNA replication machinery had the highest similarities to known genes. These include a suite of genes whose activity is directly or indirectly regulated by cyclin-dependent kinase: ribonucleotide diphosphate reductase, proliferating cell nuclear antigen (PCNA), replication factor C, replication protein A, and DNA ligase. Mitosis-specific genes include the anaphase-promoting complex protein 3 (CDC27) that controls mitosis exit through ubiquitin-directed proteolysis of M-phase-specific proteins and CDC48, an AAA ATPase that participates in spindle disassembly by targeting spindle-stabilizing proteins for proteolysis directed by anaphase-promoting complex (APC). The presence of APC-mediated activities has been reported previously in the dinoflagellate Crypthecodinium cohnii; however, no molecular or structural basis for this activity was identified (Yeung et al., 2000). Additional putative M-phase genes with lower similarity include mitotic checkpoint kinases NIMA and Bub1, and regulators of mitotic protein phosphatase 1 (Sds22), chromatid cohesion (Dif1), and chromosome condensation and segregation. Together this gene list supports the presence of a eukaryotic cell cycle machinery in dinoflagellates and provides tools for its functional analysis.
Signal Transduction Genes
The EST database also provides the first overview in dinoflagellates of signal transduction pathways present that relay environmental cues to elicit cellular responses (Table 2). Components of several conserved ser/thr protein kinase transduction cascades in eukaryotes were identified, including pathways dependant on cAMP, Ca, and calmodulin, and MAP kinase pathways. Previous studies have demonstrated the presence of cAMP-dependent kinase in two dinoflagellate species, Amphidinium operculatum (Leighfield et al., 2002) and Gonyaulax polyedra (=Lingulodinium polyedrum; Salois and Morse, 1997). A MAP kinase has also been reported in Pfiesteria piscicida (Lin and Zhang, 2003). In contrast, protein tyrosine kinases were not found, which is consistent with the notion that they diverged from ser/thr kinases with the emergence of metazoans (Kruse et al., 1997), although reversible Tyr phosphorylation was reported in Prorocentrum lima (Dawson et al., 1997). ESTs with similarity to both ser/thr protein phosphatases (type 1, type 2a, and type 2c), which oppose the actions of ser/thr kinases in signaling cascades, and dual function (ser/thr/tyr) protein phosphatases, involved in many growth-regulatory processes, were identified. The presence of type 1 and 2 ser/thr protein phosphatases in dinoflagellates has previously been demonstrated (Boland et al., 1993; Comolli et al., 1996; Sugg and Van Dolah, 1999). ESTs with moderate similarity to known membrane-bound signal receptors were identified, including an acetylcholine receptor (e−16) and opioid growth factor receptor (e−33). A cryptochrome blue light receptor revealed in K. brevis by EST analysis is the first light-dependent receptor reported in dinoflagellates.
Transcription/Translation Genes
The transcriptional and translational machinery comprises a third class of genes critical to understanding dinoflagellate gene expression, and is of particular interest because of the absence of nucleosomes typically involved in eukaryotic gene expression. Together these processes were represented by 15% of the expressed ESTs with similarity to known genes. Members of the basal transcriptional apparatus identified in the library include RNA polymerase I, which is responsible for ribosomal RNA synthesis and localized to the nucleolus. No EST with similarity to RNA polymerase II, which is responsible for eukaryotic mRNA transcription, was identified. However, the presence of RNA polymerase II in dinoflagellates is supported by their sensitivity to α-amanitin (Rizzo, 1979). The canonical promoter sequence TATA has not been found in dinoflagellates to date. However, a protein with similarity to TBP (cTBP), which shows structural similarity to TATA box-binding proteins (TBPs), but preferentially binds to TTTT, has been described in C. cohnii (Guillebault et al., 2002). The K. brevis library contains only a single EST with modest similarity to an accessory transcription initiation complex, TFIIE. Whether the absence of ESTs with similarity to genes involved in transcriptional regulation reflects divergence due to the unusual chromatin structure of dinoflagellates, or simply reflects their low level of expression, remains to be determined. A number of genes involved in RNA processing were evident, including genes involved in polyadenylation, splicing, and RNA stability. In contrast to transcription, numerous components of the translation machinery are present with good similarity to known genes (Table 3), including translation initiation and elongation factors. Of these, EIF5a has previously been reported in C. cohnii (Chan et al., 2002).
Codon Usage and GC Content
Fifty K. brevis ESTs, selected from those with the highest similarity to previously identified genes in GenBank and those having no similarity to known genes, were chosen to determine the degree of GC-rich coding regions and general information about codon usage. The average GC content in these K. brevis ESTs was 51%, with a G or C in the third position in 53.5% of codons. This is similar to the GC content of coding regions (CDS) in Amphidinium carterae (50.44% in 13 CDSs) and C. cohnii (50% in 19 CDSs); in contrast, L. polyedrum has a higher degree of GC-rich coding regions (59%) with a 75% preference for a G or C in the third position (Codon Usage Database, GenBank Release 140.0, March 2004). These findings suggest that dinoflagellates may vary widely in their use of GC in coding regions. The GC content of K. brevis coding regions is in an intermediate range between bacteria, mammalian species, and higher plants at 62%, 52%, and 45% GC content, respectively.
Prevalence of Single Nucleotide Polymorphisms
Single nucleotide polymorphisms (SNPs) are the most common type of sequence variation between gene alleles and can be used as tools in genetic mapping, estimating population diversity, and correlating genotype to phenotype. SNPs identified from EST databases are especially informative in that they identify population diversity within expressed genes (Kota et al., 2001). There is growing precedence for the presence of multiple copies of genes in dinoflagellates. The peridinin chlorophyll α–binding protein (PCP) gene family is present in 5000 copies per 200 pg of DNA in the genome of L. polyedrum (Le et al., 1997), one of the largest gene families reported for any organism. The same gene family is present in 36 copies in Symbiodinium, a symbiotic dinoflagellate associated with corals that has a substantially smaller genome of 3 pg (Reichman et al., 2003). The PCP genes are present in tandem arrays, and in Symbiodinium 89% of clones screened from genomic and cDNA libraries were distinct at the nucleotide level. Similar tandem repeats of gene families have been reported for luciferin-binding protein and rubisco (Lee et al., 1993; Machabee et al., 1994; Rowan et al., 1996). A cAMP protein kinase from L. polyedrum is also present in 30 copies (Salois and Morse, 1997). Of the 305 K. brevis contigs containing more than 2 ESTs, 39.7% contained SNPs. To determine whether a discrepant base might be a SNP or an experimental error, we relied on sequence quality information from the trace reads and replication of base calls from multiple ESTs within the contig.
Visual inspection of sequencing chromatograms or sequence alignments in EST libraries from other organisms has revealed SNPs at a frequency of occurrence of 0.82 per 100 bp in barley (Bundock et al., 2003) and 1.32 per 100 bp in catfish (He et al., 2003). In the K. brevis EST library, the estimated overall frequency of SNPs in contigs containing 10 or more ESTs was 1 in 90 bp. Of these, 85% are in the third codon position, indicating a synonymous substitution that does not alter protein sequence. The prevalence of SNPs in the expressed sequences of K. brevis suggests that multiple copies of many genes exist in this dinoflagellate. A similar frequency of occurrence of SNPs was observed in L. polyedrum and A. carterae (Bachvaroff et al., 2004).
Development of a K. brevis DNA Microarray
The availability of ESTs from K. brevis not only provides the first insight into the expressed genome of this problematic organism, but also provides the tools with which to begin investigating mechanisms regulating its growth and toxicity. In addition to the 1531 unigenes with similarity to known genes, the library contains 3749 sequences of unknown function. Therefore we have chosen to take a genomewide transcript profiling approach to characterize responses of known and unknown genes to conditions known to alter growth, cell cycle progression, and toxicity. To facilitate this, 60-bp oligonucleotides were designed to uniquely recognize gene sequences sharing identity as defined by BLASTx similarity scores and which cluster using an identity of 70% over 30 bp. The resulting oligonucleotide probes and controls were printed on glass slides yielding an 8455 feature array, including 4629 primary probes to each of the unigenes, 3462 replicate probes to selected genes, and 364 hybridization controls (Figure 5). Replicate probes were randomly distributed over the array to minimize any bias that may arise owing to probe position.
Initial experiments were carried out to optimize labeling protocols and validate probe specificity and reproducibility between replicate probes. Preliminary experiments determined that direct Cy3/Cy5 labeling of amplified RNA provided superior reproducibility to labeling of cDNA targets. Therefore the RNA amplification method was selected for use with the microarray. For probe validation a pooled sample of RNA from daytime and nighttime cultures was used in order to maximize the number of genes expressed, since genes being expressed during daytime hours, such as photosynthetic genes, may be downregulated at night, while most likely many genes expressed at night will be downregulated during the day. Of the 8091 (noncontrol) features, 90.6% produced a mean fluorescence signal that was greater than twice the average background. The mean signal-to-background ratio was 58.3. Global background intensities averaged from the 3 arrays were 50 counts in the Cy5 channel and 46 counts in the Cy3 channel. Excellent repeatability was found between duplicate probes, in terms of both fluorescence intensity and fold change in response. Thus the microarray appears to provide sufficient sensitivity and reproducibility for probing global gene expression in K. brevis.

Karenia brevis microarray. Self-versus-self hybridization using Cy3 and Cy5 labeled RNA targets on a K. brevis 60-mer oligo microarray. The inlay shows an 8 × 10 block enlargement of features.
Variability of gene expression must next be assessed for each gene on the array through multiple replications of an experiment. Once the normal range of variability is defined, it will be possible to define threshold levels above which a certain fold change in expression for a particular gene may be called significant. Parallel experimentation must be done to address whether biological significance can be inferred from changes in transcription assessable by microarray analysis. Under seemingly identical culture conditions, the level of expression for some genes varies greatly, with little physiologic impact. Conversely, minor changes in transcription of other genes may cause significant physiologic changes owing to translational or posttranslational regulation of gene expression, which has been observed in some dinoflagellate genes involved in circadian controlled bioluminescence. Our current investigations therefore focus on changes in global gene expression in K. brevis in response to two major physiologic regulators of K. brevis growth and behavior, the light-dark cycle and the circadian clock.
Conclusions
We have established a database of K. brevis ESTs from cells in logarithmic phase of growth. This is the first genomic database to be developed on a fucoxanthin-containing dinoflagellate, K. brevis, and provides the basis from which to begin functional genomic studies on this HAB species. To date, 7001-high quality ESTs have been analyzed and clustered into 5280 nonredundant groups. Cluster analysis and protein similarity searches indicated that the vast majority of genes expressed by K. brevis fall into a low-abundance class. The average GC content in these K. brevis ESTs was 51%, with a G or C in the third position in 53.5% of codons, indicating dinoflagellates may vary widely in their use of GC coding regions. Of the 305 K. brevis contigs containing more than 2 ESTs, 39.7% contained SNPs. The prevalence of SNPs in the expressed sequences of K. brevis suggests that multiple copies of many genes exist in this dinoflagellate.
We have identified for the first time in a dinoflagellate conserved eukaryotic genes involved in cell cycle control, intracellular signaling, and the transcription/translation machinery that are critical to understanding growth regulation in this organism. Despite its unusual chromatin structure and mitotic apparatus, analysis of the EST database suggests that K. brevis possesses typical components of the eukaryotic cell cycle machinery. Similarly, members of all major signal transduction cascades and the protein phosphatases that oppose them were found. In contrast, the near absence of identifiable transcription factors in the EST database, and the lack of identifiable TATA boxes in other dinoflagellate genes, may reflect some divergence in transcription factors relative to other eukaryotes.
Although 29% of the ESTs in the K. brevis library had identity with genes in the GenBank nonredundant database, 71% had little or no sequence identity to known genes. A promising approach to understanding the regulation of known genes and the function of unknown genes in an uncharacterized genome is the examination of their transcriptional responses by an array-based monitoring system. To this end, we designed 60-mer oligonucleotide probes specific for each unigene and developed an 8455 feature microarray including 4629 primary probes to each unigene, 3826 replicate probes to randomly selected genes, and 192 hybridization controls. Following quality control experiments, 90.6% of the features produced a mean florescence 2 times that of the average background and good repeatability between replicate probes. Therefore we determined the microarray was satisfactorily sensitive and specific enough to probe genomewide functional analysis of K. brevis genes. This approach will yield insight into the expression of known genes and their associated regulatory pathways in response to different exposure conditions. In addition, microarray analysis will assist in identifying gene function of the large number of K. brevis genes that are unidentifiable, by their coordinated responses with known genes in response to specific challenges. Thus we anticipate that the microarray will provide a powerful tool for investigations into the pathways that control the growth and toxicity of HABs.
References
Altschul SF, Gish W, Miller EW, Myers EW, Lipman DJ., (1990). Basic logical alignment search toolJ Mol Biol 215:403–410
Anderson DM, Grabher A, Herzog M., (1992). Separation of coding sequences from structural DNA in the dinoflagellate Crypthecodinium cohniiMol Mar Biol Biotech 1:89–96
Bachvaroff TS, Concepcion GT, Rogers CR, Herman EM, Delwiche CF., (2004). Dinoflgaellate expressed sequence tag data reveal massive transfer of chloroplast genes to the nuclear genomeProtist 155:65–78
Barbier M, Leighfield T, Soyer-Gobillard MO, Van Dolah FM., (2003). Permanent expression of a cyclin B homologue in the cell cycle of the dinoflagellate Karenia brevisJ Eukaryot Micro 50(2):123–131
Bhaud Y, Geraud M, Ausseil J, Soyer-Gobillard M, Moreu H., (1999). Cyclic expression of a nuclear protein in a dinoflagellateJ Eukaryot Microbiol 46(3):259–267
Boland MP, Taylor MFJR, Holmes CFB., (1993). Identification and characterization of a type-1 protein phosphatase from the okadaic acid producing marine dinoflagellate, Prorocentrum limaFEBS 334:13–17
Bundock PC, Christopher JT, Eggler P, Ablett G, Henry RJ, Holton TA., (2003). Single nucleotide polymorphisms in cytochrome P450 genes from barleyTheor Appl Genet 106:676–682
Chan KL, New D, Ghandhi S, Wong F, Lam CMC, Wong JTY., (2002). Transcript levels of the eukaryotic translation initiation factor 5A gene peak at early G1 phase of the cell cycle in the dinoflagellate Crypthecodinium cohniiAppl Environ Microbiol 68(5):2278–2284
Comolli J, Taylor W, Rehman J, Hastings JW., (1996). Inhibitors of serine/threonine phosphoprotein phosphatases alter circadian properties in Gonyaulax polyedraPlant Physiol 111:285–291
Dawson JF, Ostergaard HL, Klix H, Boland MP, Haoles CFB, (1997). Evidence for reversible tyrosine phosphorylation in the okadaic acid–producting dinoflagellate Prorocentrum limaJ Eukaryot Microbiol 44:89–95
Fast NM, Xue L, Bingham S, Keeling P., (2002). Re-examining aveolate evolution using multiple protein molecular phylogeniesJ Eukaryot Microbiol 49(1):30–37
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B., (2002). Genome sequence of the human malaria parasite Plasmodium falciparumNature 419: 498–511
Geesey ME, Tester PA (1993) Gymnodinium breve: ubiquitous in Gulf of Mexico surface waters. In: Toxic Phytoplankton Blooms in the Sea, Proceedings of the Fifth International Conference on Toxic Marine Phytoplankton, Newport, Rhode Island, U.S.A, October 28 –November 1, 1991, Smayda TJ, Shimizu Y, eds. (Amsterdam: Elsevier Science Publishers), pp 251–25
Guillard RRL. (1973). Division rates. In: Stein J.R., (ed). Handbook of Phycological Methods — Culture Methods and Growth Measurements, Cambridge, U.K.: Cambridge University Press, 289–311
Guillebault D, Sasorith S, Derelle E, Wurtz JM, Lozano JC, Bingham S, Tora L, Moreau H. (2002). A new class of transcription initiation factors, intermediate between TATA box-binding proteins (TBPs) and TBP-like factors (TLFs), is present in the marine unicellular organism, the dinoflagellate Crypthecodinium cohniiJ Biol Chem 277(43):40881–40886
He C, Chen L, Simmoms M, Li P, Kim S, Liu ZJ., (2003). Putative SNP discovery in interspecific hybrids of catfish by comparative EST analysisAnim Genet 34:445–448
Jenny M, Ringwood AH, Lacy E, Lewitus AJ, Kempton JW, Gross PS, Warr GW, Chapman RW., (2002). Potential indicators of stress response identified by expressed sequences tag analysis of hemocytes and embryos from the American oyster, Crassostrea virginicaMar Biotechnol 4:81–93
Kamykowski D, Milligan EJ, Reed RE., (1998). Biochemical relationships in the orientation of the autotrophic dinoflagellate Gymnodinium breve under nutrient replete conditionsMar Ecol Prog Ser 167:105–117
Kim YS, Martin DF. (1974). Effects of salinity on synthesis of DNA, acidic polysaccharide and ichthyotoxin in Gymnodinium brevePhytochemistry 13:533–538
Kota R, Kumar VR, Thiel T, Dehmer K. J, Graner A., (2001). Generation and comparison of EST-derived SSRs and SNPs in barley (Hordeum vulgare L.)Hereditas 135:145–151
Kruze M, Muller IM, Muller WG., (1997). Early evolution of metazoan serine/threonine and tyrosine kinases: identification of selected kinases in spongesMol Biol Evol 14:1326–1334
Le QH, Markovic P, Hastings JW, Jovine RVM, Morse D. (1997). Structure and organization of the peridinin chlorophyll protein gene in Gonyaulax polyedra. Mol Gen Genet 255:595–604
Lee DH, Mittag M, Sczelan S, Morse D, Hastings J.W., (1993). Molecular cloning and genomic organization of a gene for luciferin-binding protein from the dinoflagellate Gonyaulax polyedra J Biol Chem 268:8842–8850
Leighfield TA, Barbier M, Van Dolah FM., (2002). Evidence for cAMP-dependant protein kinase in the dinoflagellate, Amphidinium operculatumComp Biochem Physiol 133B(3):317–324
Lew DJ, Dulic V, Reed SI., (1991). Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeastCell 66:1197–1206
Lin S, Zhang H., (2003). Mitogen-activated protein kinase in Pfiesteria piscicida and its growth rate-related expressionAppl Environ Microbiol 69(1):343–349
Machabee S, Wall L, Morse D., (1994). Expression and genomic organization of a dinoflagellate gene familyPlant Molec Biol 25:23–31
Miller-Morey JS, Van Dolah FM., (2004). Differential responses of stress proteins, antioxidant enzymes, and photosynthetic efficiency to physiological stresses in the Florida red tide dinoflagellate, Karenia brevisComp Biochem Physiol C 138(4):493–505
Reichman JR, Thomas W, Peter V., (2003). PCP gene family in Symbiodinium from Hippopus hippopus: low levels of concerted evolution, isoform diversity, and spectral tuning of chromophoresMol Biol Evol 20(12):2143–2154
Rizzo PD., (1982). Isolation and properties of isolated nuclei from the Florida red tide dinoflagellate Gymnodinium breveJ Protozool 29:217–222
Rizzo PJ., (1979). RNA synthesis in isolated nuclei of the dinoflagellate Crypthecodinium cohniiJ Protozool 26:290–294
Rowan R, Whitney SM, Fowler A, Yellowlees D., (1996). Rubisco in marine symbiotic dinoflagellates: form II enzymes in eukaryotic oxygenic phototrophs encoded by a nuclear multigene familyPlant Cell 8:539–553
Salois P, Morse D., (1997). Characterization and molecular phylogeny of a protein kinase cDNA from the dinoflagellate GonyaulaxJ Phycol 33:1063–1072
Santos SR, Coffroth MA., (2003). Molecular genetic evidence that dinoflagellates belonging to the genus Symbiodinium Freudenthal are haploidBiol Bull 204:10–20
Sigee DC., (1984). Structural DNA and genetically active DNA in dinoflagellate chromosomes. Biosystems 16:203–210
Snyder RV, Gibbs PDL, Palacios A, Abiy L, Dickey R, Lopez JV, Rein KS., (2003). Polyketide synthase genes from marine dinoflagellatesMar Biotechnol 5(1):1–12
Sugg L, Van Dolah FM., (1999). No evidence for an allelopathic role of okadaic among ciguatera associated dinoflagellates. J Phycol 35:93–108
Triplett EL, Govind NS, Roman SJ, Jovine RVM, Prezelin BB., (1993). Characterization of the sequence organization of DNA from the dinoflagellate Heterocapsa pygmaea (Glenodinium sp.)Mol Mar Biol Biotechnol 2(4):239–245
Van Dolah FM, Leighfield TA., (1999). Diel phasing of the cell cycle in the Florida red tide dinoflagellate, Gymnodinium breveJ Phycol 35S:1404–1411
Walker LM., (1982). Evidence for a sexual cycle in the Florida red tide dinoflagellate Ptychodiscus brevis (=Gymnodinium breve)Bioscience 32:809–810
Wong JTY, Leveson A, Wong F., (1997). Cyclins in a dinoflagellate cell cycle Mol Mar Biol Biotechnol 6(3):172–179
Yeung PKK, New D, Leveson A, Yam C, Poon P, Wong J., (2000). The spindle checkpoint in the dinoflagellate Crypthecodinium cohniiExp Cell Res 254:120–129
Acknowledgments
We thank Jeral Tyler and Bennie Haynes for laboratory assistance, Jeanine Miller-Morey for culture maintenance, and Paul Gross for use of the Qiagen Biobot 9600 system. EST sequencing was performed by Seqwright, Houston, Texas. Funding for this project was provided by NOAA programmatic funding for Marine Biotoxins and ECOHAB grants ECO-01-242 and NA03NOS478 to F.M.V.D.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lidie, K.B., Ryan, J.C., Barbier, M. et al. Gene Expression in Florida Red Tide Dinoflagellate Karenia brevis: Analysis of an Expressed Sequence Tag Library and Development of DNA Microarray. Mar Biotechnol 7, 481–493 (2005). https://doi.org/10.1007/s10126-004-4110-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-004-4110-6