
Abstract
A simplified technique was developed for DNA sequence-based diagnosis of harmful dinoflagellate species. This protocol integrates procedures for DNA extraction and polymerase chain reaction (PCR) amplification into a single tube. DNA sequencing reactions were performed directly, using unpurified PCR products as the DNA template for subsequent sequencing reactions. PCR reactions using DNA extracted from single cells of Cocodinium polykrikoides and Alexandrium catenella successfully amplified the target ribosomal DNA regions. DNA sequencing of the unpurified PCR products showed that DNA sequences corresponded to the expected locus of ribosomal DNA regions of both A. catenella and C. polykrikoides (each zero genetic distance and 100% sequence similarity). Using the protocol described in this article, there was little DNA loss during the purification step, and the technique was found to be rapid and inexpensive. This protocol clearly resolves the taxonomic ambiguities of closely related algal species (such as Alexandrium and Cochlodinium), and it constitutes a significant breakthrough for the molecular analysis of nonculturable dinoflagellate species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Harmful algal blooms (HABs) are a worldwide problem in the marine environment and have, in many cases, been linked to the presence of the toxic dinoflagellate genera Cochlodinium and Alexandrium. Traditionally species identification and monitoring of dinoflagellate populations has relied on microscopic examination of samples collected from wild populations. However, these conventional microscopic methods, based on the measurement of morphologic characters such as cell size and shape or the position of chloroplasts, are time-consuming and require considerable taxonomic experience (Godhe et al., 2001). In addition, many morphologic features are known to vary in response to changing environmental conditions, as well as during different growth stages (Sako et al., 1990), and are therefore considered unreliable for correct taxonomic identification.
The development of polymerase chain reaction (PCR) techniques has been a major step forward in resolving some of the taxonomic ambiguities among the dinoflagellates, owing to their simplicity and high specificity (Godhe et al., 2001). However, PCR methods generally require the use of purified genomic DNA for the PCR template. Extraction and purification requires large quantities of cells free from contaminating organisms. This represents a special challenge for species that cannot be cultured under laboratory conditions, and in such circumstances representative material must be collected from wild populations when seasonal blooms occur (Marín et al., 2001). In addition, loss of DNA occurs in many of the procedures developed for isolation of DNA from dinoflagellate.
For these reasons Marín et al. (2001) designed a method to obtain pure DNA for PCR from small numbers of dinoflagellate cells (and even from single cells). Their protocol was used to prepare genomic DNA by a combination of physicochemical and enzymatic procedures, using cells embedded in agarose beads. Bolch (2001) also reported a PCR assay for genetic identification of dinoflagellates using DNA extracted directly from single cysts and vegetative cells. This procedure used liquid nitrogen to lyse cells. Both of these techniques were found to be useful for DNA extraction and subsequent PCR amplification from single cells without loss of DNA; however, no direct DNA sequencing, using unpurified PCR products amplified from a single cell, was tested. The 2 protocols for DNA extraction and sequence analysis described above require several steps of conventional DNA cloning of the PCR products prior to sequencing. These techniques are unsuitable for routine sequence-based identification of dinoflagellate species from natural populations owing to the time and labor required for purification.
In this report we describe an advanced technique for single-cell DNA extraction and direct PCR amplification of ribosomal DNA from the harmful dinoflagellates Cochlodinium polykrikoides and Alexandrium catenella. Because DNA sequencing can be performed directly on the PCR products, this technique is time-saving and has several potential applications for the analysis of genetic diversity within natural HAB populations.
MATERIALS AND METHODS
Cultures and Strains
Clonal culturesof Cochlodinium polykrikoides (CCPK06) and Alexandrium catenella (AxCt_K01) were kindly provided by Dr. M. Chang of the Korean Ocean Research and Development Institute. The strains were maintained in f/2 medium, pH 8.2 (Guillard and Ryther, 1962), at 15°C under a light and dark regimen of 12 hour each, with an incident light intensity per second of approximately 100 μmol photons/m2.
Single-Cell Isolation and Washing
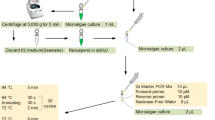
Six drops of TE buffer were deposited on a round glass petri dish with a radius of 80 cm. One drop of culture was placed in the center of the petri dish, and individual dinoflagellate cells were isolated using a sterile micropipette under a dissecting microscope (Carl-Zeiss Ltd.). Individual dinoflagellate cells were then transferred to sterile TE buffer droplets 3 to 6 times to facilitate the removal of contaminants. Individual cells, suspended in approximately 1 μl of TE buffer, were then placed in 200-μl thin-walled PCR tubes containing a drop of mineral oil. Samples were then frozen at–20°C until analysis.
Enzymatic DNA Extraction
Proteinase K (1 μl 200 μg/ml) was then added, and the tubes were maintained at 55°C for 50 minutes with a UNO-II Thermoblock (Biometra). Samples were then incubated at 95°C for a further 10 minutes to inactivate the proteinase K and facilitate DNA denaturation. The tubes were then cooled to 4°C in preparation for PCR amplification.
PCR Amplification and DNA Sequencing
PCR primers based on conserved sequences among related dinoflagellate species were designed and synthesized. The primers, based on the small subunit (SSU) rRNA genes were as follows: AT18F01 (5′-CACCTGGTTGATCCTGCCAGTAG-3′) and AT18R02 (5′-GTTTCAGCCTTGCGACCATACTCC-3′). All of the primers used in this study were synthesized on a 10-column DNA synthesizer (PolyGen).
PCR reactions using the crude lysates extracted from single dinoflagellate cells were performed. Eight microliters of PCR master mix (0.5 U Taq polymerase (Promega Corp.), 0.25 mM each of all 4 dNTPs, 1 × reaction buffer, and 1.5 mM MgCl2) including 10 pmol of each primer were added to the PCR tube containing approximately 2 μl of the lysate at 4°C. Thermocycling was as follows: initially 95°C for 5 minutes, followed by 40 cycles of 95°C for 20 seconds, 52°C for 30 seconds, and 72°C for 60 seconds. After the completion of the cycles, extension was facilitated at 72°C for 6 minutes. Three microliters of PCR product was loaded onto a 1.5% agarose gel in 1 × TBE buffer (Sambrook and Russell, 1989) along with 3 μl of loading buffer. Agarose gels were stained with ethidium bromide and photographed according to standard methods (Sambrook and Russell, 1989).
DNA cycle sequencing reactions were performed directly, using PCR products as the template, without purification of the PCR-amplified fragments. The PCR products (3–6 μl) were subjected to DNA cycle sequencing using a ThermoSequenase Version 2.0 Cycle Sequencing Kit (USB) in the presence of 1.5 pmol of SAT18F01 (5′-CCAGTAGTCATATGCTTGTC-3′) nested within the PCR primers. The sequencing primer was labeled with IRD at the 5′ end. The 4 base-specific reactions were subjected initially to 94°C for 1 minute, followed by 40 cycles consisting of 94°C for 20 seconds, 50°C for 30 seconds, and 72°C for 60 seconds in the UNO-II Thermoblock. When complete, reactions were stopped by adding 4 μl of IR2 stopper loading buffer (LiCor), and the products were heat-denatured and analyzed on a model 4200 Dual Dye Automated Sequencer according to the manufacturer’s instructions.
DNA Sequence-Based Diagnosis
For sequence-based species identification, reference sequences of the SSU rRNA gene from the related species (Alexandrium for AxCt_K01, Gymnodinium and Gyrodinium for CCPK06), including the strains of both AxCt_K01 and CCPK06, were compiled privately. Nucleotide sequence analyzed from the test species was added to the reference sequences, and their sequences were aligned using CLUSTAL W with the default settings for gap inclusion and extension (Thompson et al., 1994). Molecular diagnosis of the unidentified dinoflagellates was performed with both genetic distance estimated by the Kimura 2-parameter model (Kimura, 1980) and similarity scores by GenDoc software between pairs of algal species from the aligned sequence data.
RESULTS AND DISCUSSION
The preservation of HAB species is an important consideration for the extraction of DNA from single cells and subsequent PCR assays because some preservatives act as inhibitors of enzymatic reactions (Wilson, 1997). In previous studies preservatives such as formaline and Lugoil’s iodine were found to interfere with DNA extraction and PCR amplification (Marín et al., 2001). The preservation reagent we used was a mixture of TE buffer and mineral oil in a PCR tube, to prevent evaporation and DNA contamination from other biological sources. The single dinoflagellate cells were stored at −20°C for several months without any degradation of DNA.
For some organisms whole cells can be used for PCR amplification, without the need for DNA isolation, since cell lysis can easily be achieved by heat. However, the cellulose shells of thecated dinoflagellates maintain their shape and resist cell lysis under the application of heat and some chemicals (e.g., sodium dodecylsulfate [SDS] and dithiothreitol [DTT]). Treatment of single cells with proteinase K, as described in this study, was found to be a rapid and effective method for the extraction of PCR-ready DNA from dinoflagellate cells.
The results of the direct PCR reaction using extracts taken from single cells are shown in Figure 1. PCR products amplified with AT18F1 and AT18R2 primers, and the lysates of both Alexandrium catenella and Cochlodinium polykrikoides yielded a fragment of the expected size of 1136 bp. The rRNA gene has been PCR-amplified more successfully than single-copy genes because the eukaryotic nuclear rDNA is tandemly organized, with copy numbers up to the order of 104 (Schlötterer, 1998). In previous work Bowers et al. (2000) demonstrated that the sensitivity of PCR to amplify the rRNA gene for the dinoflagellate Pfiesteria picicida was 0.6 cell. This suggested that the rRNA gene was a useful region for PCR amplification from single cells isolated from natural samples.

PCR products amplified from single-cell DNA extracts from Cochlodinium polykrikoides (lane 1) and Alexandrium catenella (lane 2) using AT18F1 and AT18R2 primers, respectively, for the ribosomal DNA region, with 100-bp ladder size marker (lane M). The size of the fragment in lanes 1 and 2 was 1136 bp.
The techniques for single-cell PCR also have been used in the detection of the HIV-1 genome (Bertram, et al., 1995), and in PCR analyses of single fly (Gloor et al., 1993) and dinoflagellate cells (Bolch, 2001; Marín et al., 2001). The method used here simplifies several steps, as compared with previous protocols (Bolch, 2001; Marín et al., 2001). In addition, this technique allows direct DNA sequencing using unpurified PCR products amplified from single cells in order to avoid problems caused by the false-positive and false-negative signals possibly produced in the PCR detection.
In sequencing reactions, when a DNA band of the expected size can be seen on a gel, the concentration of DNA is greater than 50 ng/μl, since a 50-ng band of double-stranded DNA generally can be detected with ethidium bromide concentrations as low as 0.5 to 1 μg/ml. The PCR products therefore were used directly as the DNA template for DNA sequencing without further check on the DNA concentration. When the band could not be visualized with ethidium bromide, or when its signal was weak, the PCR protocol was adjusted to increase the primer concentration or the number of cycles from 35 to 45. These modifications in PCR strategy efficiently improved the yield of copy DNA but also resulted in the presence of nonspecific amplified fragments. These unexpected by-products were, however, negligible for DNA sequencing reactions in the present study, possibly because of the selectivity of the nested sequencing primers and low background fluorescence in the IRD (Marziali and Aleson, 2001).
The high-quality sequence ladders of the SSU rRNA genes obtained using the SAT18F01 primer are shown in Figure 2. These sequences matched those of the preanalyzed SSU rRNA genes from the strain of AxCt_K01 (accession number AY347309) and CCPK06 (AY347309). Thus DNA sequencing of the unpurified PCR products resulted in sequences corresponding to the expected locus of rDNA of the dinoflagellate species Alexandrium catenella and Cochlodinium polykrikoides.

Data obtained by direct sequencing of unpurified PCR products from SSU rRNA genes of Cochlodinium polykrikoides (A) and Alexandrium catenaella (B). Sequences show over 100 bp from the primer. The 4 lanes in each panel are in the order A-T-G-C.
For sequence-based diagnosis the partial sequence of the SSU rRNA genes from the clone AxCt_K01 was added to the reference sequences, which each consisted of about 600 bp of the partial SSU rDNAs, and the compiled sequences were aligned using CLUSTAL W software. The partial sequence of the SSU rRNA genes from the clone AxCt_K01 was zero genetic distance and 100% sequence similarity with a strain of YSC9811, obtained from GenBank (accession number AB088334). The results therefore showed that the strain of AxCt_K01 was correctly identified as the toxic dinoflagellate Alexandrium catenella. In like manner the partial sequence of the SSU rRNA gene from the clone CCPK06 was identical to that of the SSU rDNA from GenBank (Cochlodinium polykrikoides; AY347309), and the homology between them was found to be 100% sequence similarity and zero genetic distance (Table 1). The results suggest that direct DNA sequencing using the unpurified PCR products from single cells is useful for sequence-based genetic identification and clearly resolves the taxonomic ambiguities of closely related algal species (such as Alexandrium and Cochlodinium).
The new approach described here has been applied for the species diagnosis of single cells isolated from field samples. The HAB samples were collected from the coastal waters of Korea for the genera Alexandrium, Gymnodinium, Gyrodinium, and Cocnlodinium, and at Juam reservoir in Korea for a freshwater dinoflagellate, Peridinium, when seasonal blooms took place. PCR reactions successfully amplified the target rDNA regions, using primer sets (AT18F1 and AT18R2 primers) and DNA extracts of single cells (Figure 3). For sequence-based diagnosis of the cells, the nucleotide sequences analyzed from the PCR products were compared with the reference sequences of SSU rDNA from the marine dinoflagellates, including a freshwater Peridinium. The dinoflagellate cells at the genus level were identified to the species level, as judged by sequence similarity (Table 2). A relatively high degree of sequence homology (>99.2%) between the revealed and the matched sequence was recorded for A. tamarense, A. catenella, C. polykrikoides, G. catenatum, and G. sanguineum, respectively. The genetic variations, which might be caused by the large geographic separation of the same species, were significantly lower at the intraspecies level than at the interspecies level (see Table 2), and the values did not affect the identification of Alexandrium and Gymnodinium cells collected from the coastal area of Korea. All the cells were identified phenotypically, and the results were concordant with the sequence-based species identification.

PCR products amplified from DNA extracts of single cells isolated from field samples using AT18F1 and AT18R2 primers for the ribosomal DNA region. Lanes 1–6, unidentified armoured dinoflagellate (genus Alexandrium); lanes 7–11, unidentified unarmoured dinoflagellate (genera Cochlodinium and Gymnodinium); lane 12, unidentified freshwater dinoflagellate (genus Peridinium); lane M, 100-bp ladder size marker. The size of the fragments was around 1100 bp.
Mainly on the basis of sequence data, the cell from Juam Reservoir was identified as P. bipes, as judged by 98.3% sequence similarity with a strain of P. bipes (accession number AF231805). The cell, however, had not been identified phenotypically as P. bipes, since the genus Peridinium contained at least 220 species, and their thecal formula and plate shape, which were used as taxonomic characters, were quite difficult to observe under the light microscope. Furthermore, the data available in public resources to use as reference sequences for each Peridinium were not sufficient to discriminate their taxonomy.
As we described above the sequence-based diagnostic method is dependent on sequence comparison with the prerevealed sequence for species identification. The method therefore could be applied to identify several species previously known to be different, but it, also was applied to identify an unpresented dinoflagellate or HAB species from a studied area when nucleotide sequences of the same genomic regions (e.g., 18S rDNA sequence) had been reported in public resources. To date the nucleotide sequences from the HAB species, except for several Alexandrium and Gymnodinium species, have not been reported sufficiently. For the wide application of this method, further studies are needed to determine the nucleotide sequences of the rRNA and other genes of more samples collected from local or different geographic regions. In the present study we demonstrated the applicability of sequence-based diagnosis from single dinoflagellate cells, including nonculturable species.
The method described here is a rapid and inexpensive way to the isolate and preserve dinoflagellate DNA. This protocol constitutes a significant breakthrough in the application of PCR techniques to many nonculturable dinoflagellates isolated from natural samples. In addition, it is suitable for molecular diagnosis (e.g., sequence-based typing and the detection of infectious disease) and large-scale DNA sequencing of the same genomic regions from related species for molecular evolution studies.
References
S. Bertram F.T. Hufert D. Neumann-Haefelin D Laer Particlevon (1995) ArticleTitleDetection of DNA in single cells using an automated cell deposition unit and PCR Biotechniques 19 616–620 Occurrence Handle1:CAS:528:DyaK2MXos1Krt74%3D Occurrence Handle8777056
C.J.S. Bolch (2001) ArticleTitlePCR protocols for genetic identification of dinoflagellates directly from single cysts and plankton Phycologia 40 162–167
H.A. Bowers T. Tengs H.B Glasgow J.M. Burkholder P.A. Rublee D.W. Oldach (2000) ArticleTitleDevelopment of real-time PCR assays for rapid detection of Pfiesteria piscicida and related dinoflagellates Appl Environ Microbiol 66 4641–4648 Occurrence Handle1:CAS:528:DC%2BD3cXnvFyrsLg%3D Occurrence Handle11055905
A. Godhe S.K. Otta A.S. Rehnstam-Holm I. Karunasagar I. Karunasagar (2001) ArticleTitlePolymerase chain reaction (PCR) based detection of Gymnodinium mikimotoi and Alexandrium minutum in field samples from Southwest India Mar Biotechnol 3 152–162 Occurrence Handle1:CAS:528:DC%2BD3MXksVGmsr4%3D Occurrence Handle14961378
R.R.L. Guillard J.H. Ryther (1962) ArticleTitleStudies of marine planktonic diatoms, 1: Cyclotella nana Hustedt, and Detonula confervacea (Cleve) Gran Can J Microbiol 8 229–239 Occurrence Handle1:CAS:528:DyaF38XktlWqu70%3D Occurrence Handle13902807
M. Kimura (1980) ArticleTitleA simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences J Mol Evol 16 111–120 Occurrence Handle1:CAS:528:DyaL3MXmtFSktg%3D%3D Occurrence Handle7463489
I. Marín A. Aguilera B. Reguera J.P. Abad (2001) ArticleTitlePreparation of DNA suitable for PCR amplification from fresh or fixed single dinoflagellate cells Biotechniques 30 88–93 Occurrence Handle11196325
Y. Sako C.H. Kim H. Ninomiya M. Adachi Y. Ishida (1990) Isozyme and cross analysis of mating populations in the Alexandrium catenella/tamarense species complex E. Granili B. Sundstrom L. Eldler D.M. Anderson (Eds) Toxic Phytoplankton Blooms in the Sea Elsevier Amsterdam, Netherlands 95–102
J. Sambrook D.W. Russell (1989) Molecular Cloning: A Laboratory Manual Cold Spring Harbor Press New York, N.Y. 54–517
C. Schlötterer (1998) Ribosomal DNA probes and primers A. Karp P.G. Isaac D.S. Ingram (Eds) Molecular Tools for Screening Biodiversity Chapman & Hall London, U.K. 267–276
J.D. Thompson D.G. Higgins T.J. Gibson (1994) ArticleTitleCLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice Nucleic Acids Res 22 4673–4680 Occurrence Handle1:CAS:528:DyaK2MXitlSgu74%3D Occurrence Handle7984417
L.G. Wilson (1997) ArticleTitleInhibition and facilitation of nucleic acid amplification Appl Environ Microbiol 63 3741–3751 Occurrence Handle1:CAS:528:DyaK2sXms1Sgsrg%3D Occurrence Handle9327537
Acknowledgments
We thank Ki-Byum Chang, Hyun Hwa Lee, and Syung Hee Hwang for DNA sequencing. We are grateful to anonymous referees for corrections and critical comments. Financial support of this work from the Fund Support Program for Maritime Small and Venture Business from the Ministry of Maritime Affaires & Fisheries (Management No. 00-01-04) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ki, JS., Jang, G.Y. & Han, MS. Integrated Method for Single-Cell DNA Extraction, PCR Amplification, and Sequencing of Ribosomal DNA from Harmful Dinoflagellates Cochlodinium polykrikoides and Alexandrium catenella. Mar Biotechnol 6, 587–593 (2004). https://doi.org/10.1007/s10126-004-1700-x
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s10126-004-1700-x