
Abstract
Background
Oncostatin M receptor (OSMR) is a member of the interleukin 6 (IL-6) receptor family that transduces signaling events of Oncostatin M (OSM). OSM–OSMR signaling plays a key role in inflammation and cancer progression. However, the role of OSM–OSMR in gastric cancer (GC) is still unknown.
Methods
OSMR expression in GC was determined by real-time PCR (RT-PCR), immunohistochemistry (IHC) and Western blot. The effects of OSM–OSMR on GC cell proliferation, migration, invasion, and epithelial–mesenchymal transition (EMT) in vitro and metastasis in vivo were examined. The pathways underlying OSM–OSMR signaling were explored by Western blot. Regulatory mechanism between SP1 and OSMR was explored in vitro.
Results
OSMR was highly expressed in GC tissues and its expression level was closely associated with age, T stage, Lauren classification, lymph node metastasis, TNM stage and worse prognosis of patients with GC. Knockdown of OSMR expression in GC cells significantly inhibited cell proliferation, migration, invasion, and EMT in vitro, as well as tumorigenesis and peritoneal metastasis in vivo induced by OSM. These effects mediated by OSM–OSMR were dependent on the activation of STAT3/FAK/Src signaling. SP1 could bind to the promoter region of human OSMR gene from − 255 to − 246 bp, and transcriptionally regulated OSMR overexpression in GC cells.
Conclusions
OSM–OSMR contributes to GC progression through activating STAT3/FAK/Src signaling, and OSMR is transcriptionally activated by SP1.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) is one of the most lethal malignances in the world, with 952,600 new cases and 723,100 deaths reported worldwide in 2012 [1]. Gastrectomy and chemotherapy are common therapeutic approaches used for patients with GC. However, the prognosis remains poor in China, with a 5-year survival rate of 35.9% in 2010–2014 [2]. This may be attributed to its highly metastatic potential and frequent recurrent incidence. Therefore, exploring the mechanisms underlying GC progression would be useful for identifying novel therapeutic targets.
OSMR, a member of the interleukin 6 (IL-6) receptor family, is a type-II Oncostatin M (OSM) receptor that is heterodimerized by OSMR beta subunit (OSMRβ) and IL-6 signal transducer gp130. OSM is a multifunctional cytokine produced mainly by activated monocytes, macrophages, T cells, dendritic cells, neutrophils, peripheral blood mononuclear cells and Schwann cell precursors [3]. Upon OSM treatment, OSMR signals through multiple pathways including JAK/STAT, ERK1/2, p38, JNK, PI3K/AKT, and PKCδ pathways [3], leading to the transcription of a wide range of context-dependent target genes. The role of OSM–OSMR signaling in cancer progression is dependent on its target organs and cells. OSM–OSMR suppresses the growth of melanoma, lung adenocarcinoma, breast cancer and glioma [4,5,6,7]. In contrast, OSM–OSMR promotes the growth of astroglioma [8] and cervical squamous cell carcinoma [9]. It has been reported that OSMR is overexpressed in GC [10]. However, the role and mechanism underlying the effect of OSM–OSMR on GC remain unknown. Therefore, discovering the significance of OSM–OSMR in GC may provide new solutions for GC treatment.
The study investigated the expression of OSMR in human GC and explored the role and molecular mechanisms of OSM–OSMR in GC. It was found that OSMR was significantly elevated in GC tissues and closely linked with age, T stage, Lauren classification, lymph node metastasis, TNM stage and short survival of patients with GC. In GC cells that overexpressed OSMR, OSM treatment promoted the proliferation and the metastatic potential in vitro and in vivo, which was impeded by knockdown of OSMR expression. Further studies revealed that the oncogenic effects mediated by OSM–OSMR signaling were through the activation of STAT3/FAK/Src pathway. SP1 could bind to the promoter region and activate the transcription of OSMR. These findings indicated that OSM–OSMR signaling plays a vital role in GC development. Disrupting the signaling of OSM–OSMR by targeting OSMR may provide a novel therapeutic strategy for the treatment of GC.
Methods
Ethical approval and consent to participate
This study was approved by the Ethics Committee of Ruijin Hospital, Shanghai, China. All the experiments performed were under the principles of the Declaration of Helsinki. Written informed consent was obtained from each patient.
Clinical samples, cell lines, and reagents
176 pairs of GC samples were collected from patients who underwent D2 gastrectomy in Ruijin Hospital between 2012 and 2018 without chemotherapy or radiotherapy. The samples collected were divided into two parts, one for formalin fixation and another stored at liquid nitrogen for protein and RNA extraction. The pathological stage was evaluated by two pathologists individually according to American Joint Committee on Cancer staging manual (7th Edition).
Gastric cancer cells (SGC-7901, BGC-823, MGC-803, MKN-45, AGS, HGC-27 and HTB-103) and immortal gastric mucosal epithelial cell line(GES-1) were purchased from Cell Bank of Chinese Academy of Sciences, and were cultured in 90% RPMI-1640 medium (Gibco, San Francisco, USA) with 10% fetal bovine serum (Gibco) in a humidified incubator. Other reagents used have been listed in Supplementary Table S1.
Plasmids construction and transfection
OSMRβ small hairpin RNA (shRNA) and negative control (NC) sequences were designed and constructed into a hU6-MCS-Ubiquitin-EGFP-IRES-puromycin plasmid. SGC-7901 and BGC-823 cells were transfected with shRNA plasmids using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. Stable clones were selected by puromycin.
Immunohistochemistry (IHC) staining
IHC staining was performed on the tissue microarray according to the previously reported protocol [11]. The staining intensity and percentage were used to score the overall tissue sections. The evaluation criteria were listed in Supplementary Materials. Staining score was calculated as follows: overall staining score = intensity score × percentage score. A final score ≤ 3 was considered as negative staining and > 3 as positive staining.
Western blot analysis
Western blot analysis was performed as described before [11]. For a detailed description, see Supplementary Materials.
Cell proliferation, migration and invasion assays
Cell proliferation assay was performed as previously described [12]. In brief, 2×103 cells/well in 200 ul medium were seeded into a 96-well plate. Cell proliferation was determined using Cell Counting Kit 8 (Dojindo, Kumamoto, Japan) with a spectrophotometer.
Cell migration and invasion assays were conducted using 8 µm transwell inserts (Millipore) coated with (invasion assay) or without (migration assay) Matrigel Basement Membrane Matrix (BD Bioscience). 1 × 105 cells suspended in serum-free RPMI-1640 medium were added to the upper chamber. FBS-conditioned RPMI-1640 medium with OSM (20uM) or PBS was added to the lower chamber. After 24 h (migration assay) or 48 h (invasion assay), tumor cells in the lower side of the chamber were fixed and stained with crystal violet and photographed. Six random fields were chosen to calculate the number of migrated or invaded cells.
Real-time PCR (RT-PCR)
Total RNA was extracted by a TRIzol reagent (Invitrogen) and cDNA synthesis was performed using Reverse Transcription System (Promega, Madison, WI, USA) following the manufacturer’s instructions. The mRNA level of OSMR was measured via SYBER Green PCR Master Mix (Applied Biosystems). The primers used have been listed in Supplementary Table S2.
Chromatin immunoprecipitation (ChIP) assay and Dual-luciferase reporter assay
ChIP assay was performed with the EZ-ChIP™ kit (Millipore) according to the instructions. For a detailed description, see Supplementary Materials. The primers used have been listed in Supplementary Table S2.
Dual-luciferase reporter assay was performed using Dual-Luciferase® Reporter Assay System (Promega). Generally, OSMR promoter or control vector was co-transfected with SP1 expressing plasmid or empty vector into 293T cells. After 36 h of transfection, the cells were digested and the luciferase activity was detected.
In vivo tumorigenesis and peritoneal dissemination
For tumorigenesis in vivo, SGC-7901/NC or SGC-7901/SH cells were injected subcutaneously into nude mice, and OSM (12.5 ng/g) or PBS was then injected into the tail vein weekly. Tumor volume was calculated by measuring tumor length and width every week. For peritoneal dissemination, 5 × 106 tumor cells were injected intraperitoneally, followed by OSM (12.5 ng/g) or PBS injection intraperitoneally weekly. All mice were sacrificed under anesthesia four weeks after GC cell injection. Tumor grafts and peritoneal nodules were weighted and observed.
Statistical analysis
All experiments were repeated at least three times. The results were shown as mean ± SEM. Pearson χ2 test was used to display the association between OSMR expression and clinicopathological characteristics. The Student’s t test and one-way ANOVA were performed to check the differences between the treated and the control groups. The two-tailed value of p < 0.05 was considered statistically significant. The survival curve of GC patients collected was performed by the Kaplan–Meier method and log-rank test. The survival curve of data from Gene Expression Omnibus (GEO), European Genome-phenome Archive (EGA) and The Cancer Genome Atlas (TCGA) was analyzed by Kaplan–Meier plotter [13]. SPSS19.0 software was used to perform the statistical analyses.
Results
OSMR is highly expressed in GC and predicts poor prognosis
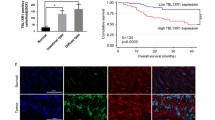
The expression level of OSMR was first determined by RT-PCR analysis in GC tissues and adjacent non-tumor tissues. Up-regulated expression of OSMR mRNA was found elevated in 66.7% (36 of 54) GC tissues than adjacent non-tumor tissues (Fig. 1a, b). IHC performed in 176 pairs of tissues showed that OSMR positive expression rate was higher in GC tissues (71.0%, 125 of 176) than adjacent non-tumor tissues (34.1%, 60 of 176). OSMR expression was mainly identified in the cell membrane and cytoplasm of GC cells (Fig. 1c, d & Supplementary Fig. S1). Moreover, the number of OSMR positive cells per field (200×) in GC tissues was significantly higher than that in adjacent non-tumor tissues (Fig. 1e). Furthermore, the protein level of OSMR in GC tissues and cell lines was significantly higher than that in non-tumor tissues or immortal gastric epithelial cell line GES-1 (Fig. 1f, g).

OSMR is highly expressed in GC and predicts poor prognosis. a, b OSMR mRNA expression was quantitated by RT-PCR and the results showed that OSMR mRNA was highly expressed in 54 GC tissues compared with non-tumor tissues. c OSMR expression in GC tissues was examined by IHC. Representative images of positive and negative OSMR expression in GC tissues and non-tumor tissues (200× and 400×). d The positive expression rate of OSMR in tumor and non-tumor tissues. e The number of OSMR positive cells in GC tissues and paired non-tumor tissues per field (200×, *p < 0.05). f OSMR expression in five pairs of GC tissues and paired non-tumor tissues was detected by Western blot. g OSMR expression in seven GC cell lines and one immortalized gastric mucosal epithelial cell line GES-1 was analyzed by Western blot. h Positive OSMR protein expression was correlated with shorter overall survival time, which was performed by Kaplan–Meier survival analysis. i The association of high OSMR mRNA expression with poor survival in total 876 patients was analyzed by Kaplan–Meier plotter
The correlation between the expression level of OSMR and clinicopathologic parameters in human GC was further examined. Pearson χ2 test showed that OSMR expression was closely related to age, T stage, Lauren classification, lymph node metastasis, and TNM stage, but not with tumor size, gender, or differentiation (Table 1). Moreover, Kaplan–Meier survival analysis showed that patients with positive OSMR expression had a shorter overall survival time than OSMR negative group (Fig. 1h). In addition, OSMR mRNA expression was significantly associated with poor prognosis in 876 patients included in GEO, EGA and TCGA databases (Fig. 1i). Together, OSMR is highly expressed in GC cells and predicts poor prognosis of GC patients.
OSM promotes the growth, migration, invasion and EMT of OSMR-overexpressing GC cells in vitro
Given the potential role of OSMR overexpression in GC progression, whether the level of OSMR expression could affect GC cell growth was first determined. OSMRβ shRNA plasmids (SH1, SH2 and SH3) were constructed and transfected into GC cells, and stable cell lines expressing SH2 plasmid were chosen for further study (Fig. 2a, b). OSM promoted the proliferation of SGC-7901 and BGC-823 cells, while knockdown of OSMR expression in GC cells attenuated the increased proliferation rate induced by OSM (Fig. 2c). These results suggest that OSMR overexpression conferred greater sensitivity to OSM in promoting GC cell growth.

OSM–OSMR promotes GC cell proliferation, migration, invasion and EMT in vitro. a, b OSMR expression in GC cells stably transfected with OSMR–SH plasmid and control vector (**p < 0.01). c The effect of OSM–OSMR on GC cell growth was detected by cell proliferation assay (*p < 0.05, **p < 0.01, ***p < 0.001). d, e The effect of OSM–OSMR on GC cell migration and invasion was detected by cell migration and invasion assays (200×). The number of migrated or invaded cells are shown (*p < 0.05). f Associations of OSMR with E-cadherin and N-cadherin expression level in the TCGA database were analyzed by LinkedOmics. g IHC staining of E-cadherin and N-cadherin in GC tissues. Representative images of OSM, E-cadherin and N-cadherin staining in GC tissues (200×). h E-cadherin and N-cadherin expression in GC patients with OSMR positive or negative expression are displayed. i, j After transfection with OSMR-SH2 plasmid or control vector, the expression of E-cadherin and N-cadherin in GC cells induced by OSM were analyzed by Western blot. j Densitometric analysis of E-cadherin and N-cadherin expression in GC cells after OSM treatment and OSMR knockdown (*p < 0.05, **p < 0.01, ***p < 0.001)
Migration and invasion are important properties of tumor cells to initiate metastasis, and the involvement of OSM–OSMR in GC cell motility was further determined. As shown in Fig. 2d, e, OSM stimulation significantly prompted the migration and invasion potential of BGC-823 and SGC-7901 cells, while knockdown of OSMR expression impaired the migratory and invasive ability of GC cells induced by OSM.
Epithelial–mesenchymal transition (EMT) is a process that provides cancer cells with enhanced motility and invasiveness. Analyses of RNA-seq data from TCGA by LinkedOmics [14] showed that OSMR expression exhibited a significant negative correlation with E-cadherin expression and a positive correlation with N-cadherin expression (Fig. 2f). In addition, IHC analysis in 130 GC tissues and paired non-tumor tissues showed that E-cadherin protein was expressed in 36.7% of OSMR+ GC (33 of 90) and 57.5% of OSMR− GC (23 of 40, Fig. 2h). N-cadherin protein was expressed in 64.4% OMSR+ GC (58 of 90) and in 37.5% OSMR− GC (15 of 40, Fig. 2h). Furthermore, OSM treatment markedly suppressed E-cadherin expression and increased N-cadherin expression, while knockdown of OSMR expression in GC cells significantly impaired this change (Fig. 2i, j). These results indicate that OSM promotes GC cell migration, invasion and EMT, which is dependent on the presence of OSMR.
The tumor-promoting effects mediated by OSM–OSMR signaling are through activating STAT3/FAK/Src pathway
Previous studies revealed that OSM mainly acts through activating STAT3 [3]. However, the downstream targets of STAT3 are cancer subtype-dependent. Focal adhesion kinase (FAK), a non-receptor protein-tyrosine kinase, plays a central role in focal adhesion assembly during cell adhesion and spreading [15]. Src family kinase, especially c-Src, has been demonstrated to play a vital role in the promotion of tumor cell motility and proliferation [16]. Western blot analysis showed that STAT3-Y705, FAK-Y397, FAK-Y576/577, FAK-Y925 and SRC-Y416 phosphorylation level was higher in GC than non-tumor tissues (Fig. 3a). OSM treatment enhanced the activation of STAT3, FAK and SRC in BGC-823 and SGC-7901 cells. However, knockdown of OSMR expression in GC cells attenuated the increased phosphorylation level of FAK, Src, and STAT3 (Fig. 3b, c). These results suggest that OSM activates STAT3, FAK, and Src, which is dependent on OSMR.

OSM–OSMR promotes GC progression via STAT3/FAK/Src signaling. a STAT3, FAK and SRC activation level was detected in three pairs of GC tissues and non-tumor tissues. b Upon OSM stimulation, the activation of STAT3, FAK and Src was analyzed by Western blot. c Densitometric analysis of p-FAK, p-STAT3 and p-Src expression (phosphorylated/total protein /GAPDH, *p < 0.05, **p < 0.01, ***p < 0.001). d Western blot analysis of the activation of STAT3, FAK and Src in GC cells treated with or without OSM, PF228 (FAK inhibitor at 10uM), STATTIC (STAT3 inhibitor at 20uM) or Saracatinib (Src inhibitor at 10uM). e Densitometric analysis of p-FAK, p-STAT3 and p-Src expression (*p < 0.05, **p < 0.01, ***p < 0.001) in GC cells treated with OSM, PF228, STATTIC or Saracatinib. f STAT3, FAK or Src inhibitor (STATTIC, PF228 and Saracatinib) attenuated the proliferation induced by OSM (**p < 0.01, ***p < 0.001). g The effect of OSM on GC cell migration and invasion was determined by cell migration and invasion assay in the presence of STAT3, FAK or Src inhibitor. Representative images of migration and invasion assay are presented. h The number of migrated or invaded cells after OSM treatment and inhibitors (PF228, STATTIC, and Saracatinib) per field (200×, **p < 0.01, ***p < 0.001). i Western blot analysis of EMT in GC cells treated with OSM and specific inhibitors. j Densitometric analysis of E-cadherin and N-cadherin expression in GC cells after treatment with OSM and specific inhibitors (**p < 0.01, ***p < 0.001)
To better understand the involvement of FAK, STAT3, and Src activation induced by OSM–OSMR in GC cells, FAK, STAT3, and Src pathways were attenuated, respectively, using specific inhibitors after OSM treatment. Inhibition of STAT3 activity by STATTIC significantly decreased the phosphorylation of FAK and Src in GC cells. In addition, the inhibition of FAK activity by PF228 also led to a striking decrease of Src and STAT3 phosphorylation. However, Saracatinib, Src-specific inhibitor, showed no effect on either FAK or STAT3 activation (Fig. 3d, e). Therefore, these data indicate that STAT3 and FAK form a positive feedback loop induced by OSM–OSMR signaling, which subsequently activates Src.
To further explore the activation of STAT3/FAK/Src pathway in mediating the tumor-promoting properties of GC cells induced by OSM–OSMR, SGC-7901 and BGC-823 cells were treated with specific FAK, STAT3, or Src inhibitor after OSM treatment, respectively. FAK inhibition abrogated the proliferation (Fig. 3f), migration, invasion (Fig. 3g, h) and EMT (Fig. 3i, j) of OSMR-overexpressing GC cells induced by OSM, so did the blockage of STAT3 activity or Src activity by their specific inhibitor STATTIC or Saracatinib. Taken together, these results suggest that the tumor-promoting effects mediated by OSM–OSMR are through activating STAT3/FAK/Src pathway.
OSM–OSMR facilitates GC cell growth and peritoneal dissemination in vivo
Whether OSM–OSMR could facilitate GC progression in vivo was further determined. SGC-7901/SH or SGC7901/NC cells were subcutaneously transplanted into nude mice, and OSM or PBS was injected via tail vein weekly. As shown in Fig. 4a, b, SGC-7901/NC cells, instead of SGC-7901/SH cells, showed aggressive proliferation in response to OSM compared with PBS treatment. Moreover, the average weight of tumors generated from SGC-7901/NC cells treated with OSM was significantly increased compared with PBS (Fig. 4c), while knockdown of OSMR expression halted the weight increase of tumors from SGC-7901/SH cells in response to OSM. Tumors generated from SGC-7901/NC cells with OSM treatment showed enhanced STAT3, FAK and SRC activation level compared with PBS treatment. While, OSMR interference attenuated the difference generated by OSM treatment (Fig. 4d). These results suggest that OSM treatment could enhance OSMR-overexpressing GC growth in vivo.

OSM–OSMR promotes GC growth and GC peritoneal dissemination in vivo. a Photographs of tumors derived from SGC-7901/NC and SGC-7901/SH cells in nude mice (N = 5 per group) injected with OSM or PBS weekly. b Subcutaneous tumor volume was measured weekly (*p < 0.05). c Weight of tumors derived from SGC-7901/NC and SGC-7901/SH cells (**p < 0.01). d STAT3, FAK and SRC expression levels were detected in the lysate of tumor tissues generated in nude mice. e Representative images of nude mice intraperitoneally inoculated with SGC-7901/NC and SGC-7901/SH cells with weekly OSM or PBS injection (N = 5 per group). f The number of nodules in the abdominal cavity of nude mice (*p < 0.05)
To further quantify the metastatic potential of GC cells overexpressing OSMR in vivo, SGC-7901/NC or SGC-7901/SH cells were intraperitoneally transplanted into nude mice, and OSM or PBS was injected into the abdominal cavity weekly. Four weeks later, there were significantly more visible peritoneal nodules generated from SGC-7901/NC cells in response to OSM compared with PBS (Fig. 4e, f). In contrast, knockdown of OSMR expression in SGC-7901 cells attenuated the increased visible peritoneal nodules induced by OSM. Together, these results indicate that OSM–OSMR promotes GC growth and peritoneal dissemination in vivo.
OSMR is transcriptionally regulated by SP1
The oncogenic transcription factor SP1 plays a vital role in many types of cancer [17]. To determine the transcriptional regulation of OSMR in GC cells, the correlation between SP1 and OSMR in GC was analyzed using TCGA data. As shown in Fig. 5a, SP1 was positively correlated with OSMR. IHC analysis on 130 GC tissues showed that OSMR expression was significantly higher in those concomitantly with SP1 expression (Fig. 5b, c). Western blot analysis showed OSMR expression level was consistent with SP1 expression (Fig. 5d). Moreover, OSMR expression was up-regulated after SP1 overexpression, and repressed after SP1 transcription inhibition or interference. (Fig. 5e–g).

OSMR is transcriptionally activated by SP1. a Associations of OSMR with SP1 expression level in GC from TCGA database were analyzed by LinkedOmics. b IHC staining of OSMR and SP1 in GC tissues. Representative images of OSMR and SP1 staining in GC tissues (200×). c The positive expression levels of OSMR in SP1+ or SP1− GC tissues are shown. d The expression levels of OSMR and SP1 in GC cells are shown. e, f & g OSMR expression in GC cells after SP1 overexpression, transcriptional inhibition or interference was detected by Western blot (*p < 0.05, **p < 0.01). h OSMR mRNA level after SP1 overexpression, inhibition or interference was detected by RT-PCR (***p < 0.001). i The binding of SP1 to OSMR promoter was analyzed by ChIP-PCR (*p < 0.05). h The activation of OSMR gene transcription was analyzed by Dual-Luciferase reporter assay (**p < 0.01, ***p < 0.001)
The promoter region of OSMR was analyzed via the JASPAR database (http://jaspar.genereg.net/) and five potential SP1 binding regions within OSMR promoter were identified (Supplementary Table S4). ChIP analysis showed that significant enrichment of SP1 ChIP DNA in OSMR promoter region was found compared with IgG isotype antibody (Fig. 5i). Dual-luciferase reporter assay showed that SP1 significantly increased the relative luciferase activity than control group by co-transfecting the full-length promoter and OSMR promoter 1# vector (Region 5 deletion), rather than promoter 2# (Region 4, 5 deletion), 3# (Region 3, 4, 5, deletion) or 4# (Region 2, 3, 4, 5 deletion) vector. This result suggested that Region 4 (− 255 to − 121 bp), containing predicted binding site #8 (− 255 to − 246 bp) and #9 (− 130 to − 121 bp), could bind to SP1. Mutation of the predicted binding site #8 or #9 was further performed. As shown in Fig. 5j, mutation of the binding site #8, instead of the binding site #9, abrogated the difference of luciferase activity between SP1 transfection group and control group. Thus, SP1 could bind to OSMR promoter at − 255 to − 246 bp and then activate the transcription of OSMR.
Discussion
This study investigated the role and clinical significance of OSMR and its ligand OSM in GC progression. It is found that OSMR was highly expressed in GC and predicted a poor outcome. OSM could enhance proliferation and metastasis of OSMR-overexpressing GC cells via STAT3/FAK/Src signaling. These findings reveal the key role of OSM–OSMR in GC progression and provide evidence for seeking new strategies for GC treatment.
The aberrant expression of OSMR has been reported in several types of cancer, including astroglioma, cervical cancer, and breast cancer [3]. Overexpression of OSMR in cervical cancer cells leads to enhanced responsiveness to OSM, which induces a pro-angiogenic phenotype and increases cell migration and invasion [9]. High OSMR expression is also observed in breast cancer where overexpression of OSMR is related to shorter recurrence-free period and lower overall survival in breast cancer [18]. Here, OSMR was frequently overexpressed in GC, and was significantly associated with age, T stage, Lauren classification, lymph node metastasis, TNM stage and worse prognosis, which suggested that OSMR may serve as a new predictor of worse prognosis in human GC.
OSM–OSMR plays an important role in inflammation and hematopoiesis, and is increasingly being recognized as an essential factor for tumor progression [19]. However, the effects of OSM–OSMR on cancer cells seem to be context- and cell type-dependent. Herein, this study investigated the effect of OSM–OSMR on GC cells. OSM was found to promote the proliferation, migration, invasion, and EMT of OSMR-overexpressing GC cells in vitro. Correspondingly, OSM–OSMR signaling could boost GC growth and peritoneal dissemination in vivo. These results indicate that OSM–OSMR signaling could function as an important contributor to GC progression. OSM is secreted by macrophage, neutrophils, T cells, and dendritic cells [3], which is observed in the solid tumor microenvironment and could provide an abundant source of OSM. However, the cellular source of OSM in GC needs to be explored in the future study.
The downstream events following OSM–OSMR are associated with the activation of multiple pathways, including phosphorylation of STAT3. This process is mediated by the C-terminal of OSMRβ subunit [3], as OSM cannot activate STAT3 in OSMR-deficient prostate cancer 22Rv1 cells [20]. However, the signaling pathways that mediate OSM–OSMR effects on GC cells have not been fully delineated. Our present data indicate that the activation of STAT3 pathway triggered by OSM is important for the tumor-promoting effects of the OSM–OSMR in GC. These observations are in agreement with previous reports that STAT3 activation is the main event induced by OSM–OSMR in cancer cells. FAK and Src are cytoplasmic non-receptor tyrosine kinases that influence cell adhesion, motility, and proliferation. FAK is activated in response to growth factor stimulation and the engagement of integrins, while its interacting partner Src is a key mediator of the processes regulated by FAK. The activation of FAK and Src has been reported in mediating tumorigenesis, metastasis or drug resistance of various types of cancer, including GC [21], breast cancer [22], and pancreatic cancer [23]. Upon activation, FAK is rapidly auto-phosphorylated at the Y-397, a binding site for Src, and Src consequently mediates the phosphorylation of FAK at the Y925 and Y576/Y577 sites. Src contains two phosphorylation sites, Y416 and Y527. The phosphorylation of Y416 site reflects promoted enzymatic activity, while the phosphorylation of Y527 decreases its activity. In this study, it is found that OSM–OSMR phosphorylated FAK at Y397, Y925 and Y576/577 sites, and Src at Y416 site in GC cells. Either FAK or Src activation could be attenuated after OSMR interference or in the presence of FAK or Src inhibitors. STAT3 inhibition led to reduced activation of FAK and Src, and FAK inhibition decreased the activation of Src and STAT3. While Src-specific inhibitor significantly dampened the phosphorylation of Src, it did not affect either FAK or STAT3 activation. Therefore, these data suggest that STAT3 and FAK form a positive feedback loop induced by OSM–OSMR, which subsequently regulates the activation of Src. However, the detailed mechanisms of FAK activation induced by STAT3 remain to be discovered in the future. In addition, the proliferation, migration, and invasion of GC cells mediated by OSM stimulation were abolished when FAK, STAT3, or Src signaling was, respectively, attenuated by their specific inhibitors, which indicated that the tumor-promoting effects induced by OSM–OSMR were mediated via the activation of the STAT3/FAK/Src pathway.
Although OSMR is important in the progression of GC, the underlying mechanisms accounting for the upregulation of OSMR expression in GC cells have not been elucidated. SP1 is an essential transcription factor in the regulation of many genes involved in multiple cellular functions such as proliferation, apoptosis, angiogenesis and migration [17]. Abnormal SP1 expression contributes to GC tumorigenesis [24]. In this study, co-expression of OSMR and SP1 in GC tissues was found. SP1 was a positive regulator of OSMR, as the overexpression of SP1 increased OSMR expression, and the suppression or interference of SP1 reduced OSMR expression. Sequence analysis revealed the presence of several putative SP1-binding sites in the promoter region of OSMR, and the results of ChIP and luciferase activity assays demonstrated that SP1 could bind to OSMR promoter at −255 bp to −246 bp and enhance its transcriptional activity. These data collectively suggest that SP1 transcriptionally activates OSMR expression in GC cells.
Recently, a small molecule heterodimerized by mouse OSMR ligand-binding domain and gp130 has been used to attenuate the pathology of inflammatory bowel disease in mouse models [25]. Our study provides evidence of blocking the tumor-promoting effect of OSM–OSMR in GC via targeting OSMR.
In conclusion, OSMR overexpression is an effective predictor of worse prognosis in human GC, and the signal triggered by OSM contributes to GC progression through the activation of STAT3/FAK/Src pathway. Moreover, OSMR could be transcriptionally activated by transcription factor SP1. Thus, our results suggest that OSMR could be a potential biomarker for clinical prognosis of GC and targeting OSM–OSMR signaling may serve as a complementary approach to prevent the progression of GC.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
Yang L, Zheng R, Wang N, Yuan Y, Liu S, Li H, et al. Incidence and mortality of stomach cancer in China, 2014. Chin J Cancer Res. 2018;30(3):291–8.
Hermanns HM. Oncostatin M and interleukin-31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev. 2015;26(5):545–58.
Kortylewski M, Heinrich PC, Mackiewicz A, Schniertshauer U, Klingmuller U, Nakajima K, et al. Interleukin-6 and Oncostatin M-induced growth inhibition of human A375 melanoma cells is STAT-dependent and involves upregulation of the cyclin-dependent kinase inhibitor p27/Kip1. Oncogene. 1999;18(25):3742–53.
Pan CM, Wang ML, Chiou SH, Chen HY, Wu CW. Oncostatin M suppresses metastasis of lung adenocarcinoma by inhibiting SLUG expression through coordination of STATs and PIASs signalings. Oncotarget. 2016;7(37):60395–406.
Douglas AM, Grant SL, Goss GA, Clouston DR, Sutherland RL, Begley CG. Oncostatin M induces the differentiation of breast cancer cells. Int J Cancer. 1998;75(1):64–73.
Halfter H, Lotfi R, Westermann R, Young P, Ringelstein EB, Stogbauer FT. Inhibition of growth and induction of differentiation of glioma cell lines by oncostatin M (OSM). Growth Factors. 1998;15(2):135–47.
Lim SY, Ahn SH, Park H, Lee J, Choi K, Choi C, et al. Transcriptional regulation of adrenomedullin by oncostatin M in human astroglioma cells: implications for tumor invasion and migration. Sci Rep. 2014;4:6444.
Caffarel MM, Coleman N. Oncostatin M receptor is a novel therapeutic target in cervical squamous cell carcinoma. J Pathol. 2014;232(4):386–90.
Junnila S, Kokkola A, Karjalainen-Lindsberg ML, Puolakkainen P, Monni O. Genome-wide gene copy number and expression analysis of primary gastric tumors and gastric cancer cell lines. BMC Cancer. 2010;10:73.
Zhou Q, Wang X, Yu Z, Wu X, Chen X, Li J, et al. Transducin (beta)-like 1 X-linked receptor 1 promotes gastric cancer progression via the ERK1/2 pathway. Oncogene. 2017;36(13):1873–86.
Wang X, Zhou Q, Yu Z, Wu X, Chen X, Li J, et al. Cancer-associated fibroblast-derived Lumican promotes gastric cancer progression via the integrin beta1-FAK signaling pathway. Int J Cancer. 2017;141(5):998–1010.
Szasz AM, Lanczky A, Nagy A, Forster S, Hark K, Green JE, et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget. 2016;7(31):49322–33.
Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46(D1):D956-D63.
Lee BY, Timpson P, Horvath LG, Daly RJ. FAK signaling in human cancer as a target for therapeutics. Pharmacol Ther. 2015;146:132–49.
Patel A, Sabbineni H, Clarke A, Somanath PR. Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci. 2016;157:52–61.
Beishline K, Azizkhan-Clifford J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015;282(2):224–58.
West NR, Murphy LC, Watson PH. Oncostatin M suppresses oestrogen receptor-alpha expression and is associated with poor outcome in human breast cancer. Endocr Relat Cancer. 2012;19(2):181–95.
Tanaka M, Miyajima A. Oncostatin M. a multifunctional cytokine. Rev Physiol Biochem Pharmacol. 2003;149:39–52.
Godoy-Tundidor S, Cavarretta IT, Fuchs D, Fiechtl M, Steiner H, Friedbichler K, et al. Interleukin-6 and oncostatin M stimulation of proliferation of prostate cancer 22Rv1 cells through the signaling pathways of p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Prostate. 2005;64(2):209–16.
Chen Y, Xia X, Wang S, Wu X, Zhang J, Zhou Y, et al. High FAK combined with low JWA expression: clinical prognostic and predictive role for adjuvant fluorouracil-leucovorin-oxaliplatin treatment in resectable gastric cancer patients. J Gastroenterol. 2013;48(9):1034–44.
Hiscox S, Jordan NJ, Morgan L, Green TP, Nicholson RI. Src kinase promotes adhesion-independent activation of FAK and enhances cellular migration in tamoxifen-resistant breast cancer cells. Clin Exp Metastasis. 2007;24(3):157–67.
Macha MA, Rachagani S, Gupta S, Pai P, Ponnusamy MP, Batra SK, et al. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling. Cancer Lett. 2013;341(2):166–77.
Xu XW, Pan CW, Yang XM, Zhou L, Zheng ZQ, Li DC. SP1 reduces autophagic flux through activating p62 in gastric cancer cells. Mol Med Rep. 2018;17(3):4633–8.
West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med. 2017;23(5):579–89.
Acknowledgements
We thank LetPub (http://www.letpub.com) for the linguistic assistance during the preparation of this manuscript. This research was funded by National Natural Scientific Funding of China [Nos. 81572798, 81672822, 81871902 and 81871904] and Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support [20152505].
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yu, Z., Li, Z., Wang, C. et al. Oncostatin M receptor, positively regulated by SP1, promotes gastric cancer growth and metastasis upon treatment with Oncostatin M. Gastric Cancer 22, 955–966 (2019). https://doi.org/10.1007/s10120-019-00934-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-019-00934-y