
Abstract
Deep tissue imaging using two-photon fluorescence (TPF) techniques have revolutionized the optical imaging community by providing in depth molecular information at the single-cell level. These techniques provide structural and functional aspects of mammalian brain at unprecedented depth and resolution. However, wavefront distortions introduced by the optical system as well as the biological sample (tissue) limit the achievable fluorescence signal-to-noise ratio and resolution with penetration depth. In this review, we discuss on the advances in TPF microscopy techniques for in vivo functional imaging and offer guidelines as to which technologies are best suited for different imaging applications with special reference to adaptive optics.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ever since man first saw the structure within a living cell, the quest to understand various molecular and cellular processes started to develop in parallel. The past few decades have witnessed rapid advancement in optical microscopy techniques for deep tissue imaging. Technology-driven imaging of live cells and their cellular processes has shaped our recent understanding of modern-day science. However, there remain some gaps within the imaging tools and biological applications, which are yet to be bridged. The recent advances in the field of neuronal imaging have shed light on disease causing factors [1] as well as neuronal plasticity [2] and circuits [3]. The use of confocal microscopy in brain imaging can successfully provide structural details of upper layer neurons as well as of dynamic processes up to 200 nm spatial resolution and 250 nm axial resolution. However, imaging tissue of deeper regions of the brain is limited mainly by scattering of light through the tissue which is quite heterogeneous. This issue is mainly resolved using two-photon fluorescence (TPF) microscopy [4,5,6,7]. Here, tightly focused ultrafast pulsed laser light provides a high-energy density photon so that two photons can drive the electronic excitation of fluorescent molecules and emit light at shorter wavelengths [5]. TPF microscopy complemented with recent advances in optical labelling of deep tissues has become the method of choice for tissue imaging in an unprecedented manner. Due to its high fluorescence intensity and depth resolution deep tissue imaging using TPF has drawn substantial attention for the study of structure and vital functions of the brain in vivo [7,8,9,10,11,12].
Imaging deeper regions of the mammalian brain has always been a challenge due to scattering and aberration caused by the complex and heterogeneous tissue. One way of encountering it with respect to wave optics is varying aberrations in the wavefront created by the sample itself. Scientists originally proposed a concept of adaptive optics (AO) to rectify wavefront distortions in telescopes caused by the earth’s atmosphere [13,14,15,16,17,18]. If the type of distortion is known, one can apply AO to create a counter distortion to minimize the overall aberration before the image formation. More recently, deformable mirrors (DM) and liquid crystal spatial light modulators (SLM) have been incorporated along the excitation path in order to overcome the wavefront distortions [19, 20]. Depending on the type of aberration, these devices can be implemented in TPF microscopy to achieve better image resolution and depth.
Owing to its higher spatial resolution and reduced photo-bleaching, TPF has not only paved the way to understand the intracellular Ca2+ dynamics but also to measure the electrical activities of brain cells in vivo [20, 21]. Recent advances in TPF have enabled scientists to study the functional changes, such as Ca2+ transients and electrical activity, within brain cells of free behaving mammals. TPF for Ca2+ imaging has provided details of neuro-circuit changes in healthy, as well as diseased, conditions. However, due to technical limitations and sample complexity, neither TPF microscopy nor high-resolution optical imaging has been able to extend to regions deeper than 1.5 mm of the brain in awake and behaving mammals [4, 22]. Recently, the combination of optical fibre delivery of short pulses and gradient refractive index (GRIN) lens optics has enabled the implementation of laser scanning endo-microscopes which can be used for imaging deeper regions of the brain in awake animals, which was previously thought to be impossible. Also, imaging deeper regions of the brain of a free moving animal has become more feasible by the incorporation of wireless photometry technology [23].
In this review, we discuss on the various techniques supplemented with TPF microscopy shedding light on the recent advances in brain imaging.
TPF microscopy for deep brain tissue imaging
A long-standing goal of optical microscopy in neuroscience has been to develop techniques that can provide cellular, sub-cellular and spine level imaging to understand the basic principles of brain development and function [24]. Combination of fluorescent probes with light microscope allows the visualization of neuronal activities with greater spatial and temporal resolution. Dendritic spines, sub-micrometre functional units of information transfer at the postsynaptic neurons, which functions at a millisecond scale serves as an important factor for understanding the brain function and activity [25]. In order to investigate such intrinsic activities within the brain, there is a requirement of robust optical technology to capture the neuronal activity at sub-cellular or spine level. TPF serves as a promising optical tool owing to it fast optical sectioning and deep penetration capacities for brain structure and functional studies [5]. Yaseen M and colleagues investigated the brain metabolism by capturing the two-photon auto-fluorescence signal from NADH in cerebral tissue of rat. TPF microscopy was modified using single-photon counting detector and specialized software to decipher the metabolic activity from neuronal population using NADH auto-fluorescence lifetime as a biomarker [26]. This study, one of many, reveals the application of TPF microscopy in exploration of metabolic activity within neuronal cells of actively behaving mammals.
Deep tissue imaging is largely limited by specimen light scattering owing to its water, lipid, protein and nucleic acid compositions. With the aid of TPF microscopy, scientists are able to record simultaneously neuronal activities from superficial neurons thus providing tremendous information about the brain function [27,28,29]. However, there remains a limitation when going deeper into the cortical regions of the brain to record the neuronal activity from a population of neurons or a single cell. This can be compensated for by increasing the excitation power at the focus; however, high power density damages the sample and hence the resolution decreases with depth due to the tissue scattering. Neuroscientists used TPF microscopy to study layer 5 of an adult mouse somatosensory cortex which utilized regenerative amplification multi-photon microscopy (RAMM) and genetically encoded calcium indicators (see Fig. 1). Taking advantage of RAMM, they were able to image the activity in dendrites of individual deep-seated neurons [21]. Nevertheless, activity recording from neuronal populations is strictly restricted to superficial cortical layers [29, 30]. One of the major reasons for this limitation has been the attenuation of the excitation light by the deep cortical tissues. Mittmann and co-workers used a regenerative amplifier to optically amplify a subset of laser pulses in the multi-photon excitation [21]. This regenerative amplifier resulted in a train of pulses with reduced frequency but with amplified energy per pulse. Using this technique, one can image the GFP-labelled cells up to a depth of ~ 1000 μm [31]. Deep imaging in homogeneously labelled tissue suffers from another fundamental issue that is auto-fluorescence from the upper cortical layers. To overcome this limitation, scientists used sparse labelling technique with RAMM to record activity from soma as well as apical dendrites of cortical (L5) neurons in the somatosensory cortex of an adult mouse. Sparse labelling of the upper layer cortical neurons not only minimizes the non-specific labelling but also increases the image quality with RAMM. Until recently, pioneering work from Yuste’s lab incorporated two-photon imaging with two-photon optogenetic to simultaneously manipulate and image large population of neuronal cells. Yang and co-workers applied holographic approach using SLM to generate 3D pattern of an awake mice brain to study the neuronal circuits. With minimal thermal damage and average power requirements, scientists were able to stimulate over 80 cells in an average imaging volume of 480 × 480 × 150 μm3 from V1 cortical region [32]. This hybrid technique merged with AO and holographic microscopy would allow scientists to stimulate and image deeper parts of the brain to better understand the neuronal circuitry.

Two-photon fluorescence neuroimaging. Imaging L5 somatosensory cortex neurons labelled with GCaMP3. a Basic optical path of the TPF microscope and the regenerative amplifier (RegA) seeded by a pulsed Ti:Al2O3 laser source (Mira). Pulses from the source laser are sub-sampled and amplified in the RegA (not to scale) and dispersion compensated. The pulses entered the microscope at a pixel frequency of < 200 kHz, whereas the pulses entering the regenerative amplifier had a frequency of ~ 76 MHz. TPF signals are collected by a photon multiplier tube (PMT). Figure is adapted with permission from [21]
In vivo femtosecond laser–based TPF microscopy has revealed vital information on neural activity for brain function, despite its limitation in imaging events at depths greater than several micrometres from the brain surface. Kawakami et al. developed a near-infrared semiconductor laser–based picosecond optical pulse source with a wavelength of 1030 nm that directly led to a TPF microscope [33]. The penetration depth was examined by imaging H-lines in mouse (Thy1-eYFP) brains and demonstrated the successful in vivo imaging of hippocampal CA1 neurons. Ryohei et al. integrated TPF with fluorescence lifetime imaging (TPF-FLIM) to investigate the fluorescence resonance energy transfer (FRET) at high resolution in individual dendritic spines in intact tissue [34]. From TPF-FLIM measurement using Ras sensors based on FRET between fluorescent proteins, it was shown that Ras activation was a highly non-linear function of neural activity and dendritic Ca2+. Therefore, integration of multi-modal optical technology led to the investigation of multi-functional activities within the neurons.
TPF microscopy with special optics for brain imaging
Attaining high spatiotemporal resolution with neuronal network information from live behaving mammals was a major unmet technological challenge of the 1990s. Later advances in available imaging techniques made deep brain imaging in an awake and head-restrained mouse possible. Initially, for awake and head-restrained primates, scientists gained information about brain functional mapping and spatiotemporal responses from neuronal populations [22]. Recently, Weijian Zong and colleagues developed a miniature two-photon microscope (mTPF) with fast high-resolution that can resolve spine level activity in freely behaving mice [35]. In animal studies, movement-associated distortion has been always a limitation for optical imaging. This was one of the major highlights of the technique developed by Zong and colleagues where they successfully overcome motion artefacts in miniaturized TPF with higher resolution and longer stability during behavioural paradigms [35]. On the other hand, even during active behaviour, it is now possible to keep the brain displacements at modest levels (~ 1–5 μm) via stable cranium and surgical steps [18]. Contrarily, imaging head-restrained animals further limits the range of behaviours that can be explored. Virtual reality–based behavioural paradigms have proved to be better associated with head-restrained imaging, but it still limits the number of behaviour tasks [19]. The cerebral hemodynamic differences between an anaesthetized mouse and an awake mouse provided crucial intrinsic neuronal signals and this had a huge impact on the understanding of brain function, but the spatial resolution was ~ 1 mm [3] and temporal resolution approximately of a few seconds. Studies from mouse habituated in a head-restrained position but allowing free movement on an exercise ball have successfully recorded Ca2+ dynamics from individual neurons [18]. TPF of Ca2+ imaging is a powerful technique to monitor activity of distinct neurons and has proved substantial in vivo studies with cellular as well as sub-cellular resolution [30, 36]. Imaging Ca2+ transients at single-cell resolution in a freely behaving mouse was not possible until the emergence of two separate approaches; mini-epifluorescence microscopes used in conjunction with GRIN lens micro-endoscopes [37] and fibre photometry [38, 39].
Imaging deeper regions of the brain, such as the lateral hypothalamus that is 5 mm from the surface of the mouse brain, requires special optics to obtain sub-cellular resolution (see Fig. 2). Recently, researchers incorporated micro-endoscopes coupled with a GRIN lens to image deeper areas of the brain tissue in an awake mouse [40]. GRIN lenses are fine rod-like shape with a refractive index profile of near parabolic type and can easily be embedded inside the tissue. Since the GRIN lens displaces tissue volume, which is linear relative to the image depth, a GRIN lens is uniquely suitable for imaging neurons in deeply buried nuclei. Use of a GRIN lens for deep tissue imaging requires surgical expertise that adds to its failure. However, the use of a guide cannula (a thin hollow glass tube closed from one end for optical contact with the tissue) reduces the probability of the lens being damaged during surgical procedures [41]. Furthermore, by using a guide cannula (outer diametre 0.61 mm) and a GRIN lens (diametre of 0.5 mm), one can conduct deep circuit imaging at single cell level over a period of one week to one month. Widely used endoscopy methods make use of multi-core fibres (MCFs). The major advantages of MCFs over other invasive tools are as follows: (i) it is passive, i.e., it does not contain any active electronic component and (ii) it is very thin (0.3 mm for 6000 cores) [42]. However, the MCFs do not provide good resolution due to the less separation (microns) between the fibre cores that limits the imaging resolution by several micrometres. With repeated imaging, mini-epifluorescence microscopes can be used to measure somatic Ca2+ currents in genetically and spatially defined neurons in a single animal and allow the analysis of the cellular network dynamics that orchestrate behaviour as well as the transition to pathological disease states [37]. On the other hand, fibre photometry utilizes an optic fibre to measure calcium-mediated fluorescence from the soma [38] or terminal edges of the genetically modefied neurons [39, 43]. With the recent advent of these imaging techniques scientists can now combine various behavioural paradigms such as sensory tasks, elevated plus maze, tail suspension and complex learning experiments using operant chambers. Nevertheless, complex behavioural paradigms including forced swim test are not feasible with the currently available mini-epifluorescence microscopes. A study conducted on CA1 hippocampal place codes used Ca2+ imaging from a population of CA1 pyramidal neurons to determine the spatial as well as the temporal dynamics of place memory [37]. The authors combined Ca2+ imaging with behavioural tests to understand the long-term dynamics of CA1 hippocampal place codes. Using a viral vector (AAV2/5-CaMKIIα-GCaMP3) to express the Ca2+ indicator in the pyramidal cells, the investigators mounted a micro-endoscope with a GRIN lens on the surface of the skull of a freely exploring mouse. Using this technique, scientists were able to trace numerous pyramidal cells reflecting the diverse range of activities in the mouse brain over long periods (more than a week). Thus, studying Ca2+ dynamics using TPF complemented with innovative optics provides a better platform to investigate spatial information about neuronal coding and how memories are formed and stored over a period. Adaptive optics has also gained importance not only in imaging deeper regions of the brain but also in heterogeneously labelled samples. Recently, Champelovier et al. applied AO-based imaging module to rodent hippocampal pyramidal cells expressing genetically encoded GCaMP6. As mentioned previously, Ca2+ imaging in rodents is of specific interest in understanding neuronal activity; AO-based TPF microscopy was applied to image hippocampal tissues samples (depth of ~200 μm) regardless of inhomogeneous labelling of the samples [44]. On similar grounds, bidirectional neuronal perturbation followed by imaging has been recorded with greater resolution thanks to the holographic module complemented with TPF microscopy. Forli A. et al. have used a hybrid TPF microscopy to image red-shifted functional indicator and bidirectional perturbation of specific cell type using blue light-sensitive opsins in vivo [45]. Since complex neuronal networks and activity are regulated spatiotemporally in the brain, development of TPF along with holographic modules allows scientists for understanding such complex networks.

In vivo optical imaging. Optical technologies developed for in vivo imaging in freely behaving animal model. Mini-epifluorescence microscope attached to a head-mounted base plate that is centred around a micro-endoscope implanted above a brain region of interest (a). A fibre probe is implanted above the target brain region of interest to collect bulk changes in calcium-mediated fluorescence using fibre photometry (b). In vivo microendoscopic imaging (left) or fiber photometry (right) for measuring the neuronal activity under social interaction. Figure is adapted with permission from [40]
Mainly, these tools improved the imaging either by incorporating better fluorescent probes or photo-switchable molecules, caged compounds and light-activated ion channels or pumps [46]. Conversely, scientists successfully modified and improved the existing optical imaging techniques to illuminate the brain in a much more precise and specific manner. Several approaches including digital micro-mirror devices (DMDs) [47], multi-beam illumination [48], acousto-optic deflectors (AODs) [49] and liquid crystal on silicon spatial light modulators (LCOS-SLMs) [50] have been developed to improve the image quality and resolution of imaging. Of the various techniques to illuminate the brain, LCOS-SLMs holds a promising future in neuroscience. Recently, it has gained importance in brain circuit studies due to its ability to generate structured light illumination by modulating the wavefront. By finely modulating the orientation of liquid crystals within each pixel (which LCOS-SLMs are made of), the desired illumination can be achieved for imaging purposes. Initially, holographic microscopy was used mainly for correcting optical aberrations in tweezers; more recently, LCOS-SLM has been used for functional imaging in neuroscience laboratories [51]. Finally, the application of TPF microscopy is not limited to brain imaging only but has been successfully implemented in imaging different retinal cells in vivo to aid early retinal degenerative problems. Using two-photon fluorescence lifetime imaging ophthalmoscopy (FLIO), Feeks and Hunter were able to label inner retinal cells with EGFP and blood capillaries with fluorescein to determine the retinal health in early degenerative stages. Authors applied AO-based FLIO to capture images from mouse retinal cells when marked with exogenous labels to target specific cell type or molecule that can be used as an indicator [52].
Adaptive optics–based TPF microscopy
TPF microscopy serves as an important tool for in vivo tissue imaging with sub-cellular resolution; however, wavefront distortions introduced by the optical system and the sample limit the achievable fluorescence signal-to-noise ratio and resolution with penetration depth [53]. The refractive index heterogeneity of the sample, the imperfections or misalignments of the optical system as well as the refractive index mismatch between the immersion medium and the sample are possible causes of aberration in optical microscopy [54, 55]. To address this limitation, several research groups introduced new methods for creating a customized two-photon excitation microscope [49, 50, 56]. Different wavefront correction techniques are reported and applied to TPF microscopy. A typical wavefront correction system contains a Shack-Hartmann wavefront sensor (SH-WS) and a deformable mirror (DM) or a SLM to sense and correct the wavefront aberrations. In most cases, the wavefront is optimized and corrected for excitation light rather than the fluorescence emission signal directly [54, 55, 57]. However, another way of estimating the optical aberration involves the direct measure of the wavefront of the fluorescence signal using a WS and a corrective pattern is applied to the SLM just before entering the detector [58, 59]. This estimation is limited to a non-scattering sample and is not suitable for a highly scattering sample such as mouse brain or in vivo imaging. A SLM-based sensorless method could overcome the limitations of sensor-based correction. In such a case, the wavefront of the excitation beam is precisely manipulated using a computational algorithm and the optimal wavefront is determined through quantifying the properties of the acquired TPF image in a feedback loop. For all of its advantages, however, adaptive element–based TPF microscopy has limitations for neuroscience due to the image acquisition time, introduction of fluorescent fiducial markers, etc. The use of adaptive optics to correct the light distortions in TPF signal promises to greatly improve the image quality and enable researchers to see more deeply and clearly into thick biological tissues (Table 1).
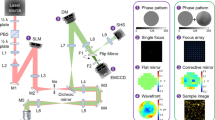
The experimental arrangement and 3D projection using wavefront sensorless adaptive optics–based TPF microscopy is described in [18, 57, 60]. An existing home-built TPF microscope was modified for AO imaging setup (Olympus BX-51). A femtosecond laser (Coherent Mira HP) was used as the excitation light source in Fig. 3 and the wavelength was tuned between 720 to 1100 nm. The laser intensity was modulated using an electro-optics device. The polarization state of light is made parallel to the active axis of SLM (Boulder Nonlinear Systems, XY-512) using a λ/2 wave-plate (Thorlabs, AHWP05M-980). The reflected beam from the SLM is Fourier-transformed and forms a first image of the target at the focal plane of achromatic lens. A telescopic combination helps in order to conjugate the SLM plane with the back focal plane of the microscope objective lens. The computer-generated hologram (CGH) modulates the phase of incident beam. The modulated light was reflected by a dichroic mirror and scans the sample through the objective lens. The fluorescence signal was collected through the excitation objective lens, by a de-scanned PMT. Details of calibration and phase mask wrapping operations can be found elsewhere [60]. Matsumoto et al. have shown that the spherical aberration in TPF microscope due to the mismatch of immersion medium and biological samples can be corrected using SLM electrically [20]. It therefore improves the fluorescence intensity and depth resolution of the observed fluorescence image. In addition, SLMs can be used in adaptive optics to correct the light path as well as sample induced aberrations using numerous optical transfer functions.

Optical layout and characteristics of 3D projection-based imaging. a The optical configuration comprises an illumination path which incorporates a SLM for 3D structured illumination and a modified imaging path using a phase mask (PM) to suppress the imaging effect of defocus. b An example of 3D illumination pattern using a 20X/0.5NA objective lens. c Ideal surface profile modulation of the phase mask. d Experimental imaging results using a transparent fluorescent slab demonstrate that the volume projection imaging path results in clearer separation of the region-of-interest signals when compared with the conventional imaging path. Note that the contrast conserves the number of photons in each image. e Wide-field imaging results comparing the conventional and volume projection imaging techniques. Note that each image is normalized to the respective peak signal. Figure is adapted with permission from [60]
AO-based TPF microscopy for brain imaging
The signal strength is reduced in 3D two-photon imaging for thick biological specimens due to sample as well as system-induced aberration [61, 62]. The ability to control and correct distorted ultra-short pulses after propagation through optical elements and a scattering medium was demonstrated [18]. The wavefront correction can be performed using a liquid crystal spatial light modulator (LC-SLM) and deformable membrane mirror membrane (DMM) (see Fig. 4). The correction can be determined using both modal and zonal approach. In zonal schemes, measurements are used to determine the optimal value at each pixel or a subset of pixels of the modulator, whereas a modal approach uses across the whole pupil and thus speeds up the optimization process [54, 55, 60]. The key to this degree of control is the optimization of a certain image metric such as brightness, sharpness or contrast of the TPF signal by iteratively adjusting the wavefront [62].

Principle of aberration correction in high-resolution optical microscopes. a Aberrations are induced as light passes through the specimen due to variations in refractive index, leading to a distortion of the focus. b The deformable mirror introduces an aberration that cancels out the specimen induced aberration, restoring a diffraction-limited focus. c The deformable mirror also corrects for aberrations induced in the detection or imaging path. Figure is adapted with permission from [67]
Genetic or hill-climbing algorithms have long been used to search for the optimal aberration correction by maximizing image brightness. The method has been used to improve the image quality in TPF microscopy of ex vivo ocular tissue as well as astrocytes in mouse brain in vivo [18, 63]. Marsh et al. has shown that by optimizing the aberration using feedback from TPF intensity of a single point in the sample allowed for the improvement of the axial resolution across the whole field of view of the image at a fixed sample depth [64]. The non-linear TPF process was incorporated with a deformable mirror for accurate measurement of aberration and is sensitive to both spatial focusing and the temporal duration of the exciting field. However, Debarre et al. demonstrated that a modal sensorless AO approach increased the brightness and contrast of TPF microscopy in mouse embryo and fixed brain slices [65, 66]. In this method, a quality metric was calculated using the intensity (mean pixel value) of the image upon sending a set of 11 low-order Zernike modes to the deformable mirror. Booth et al. has shown that by using orthogonal functions (Zernike polynomials), the aberration can often be accurately represented by a short sequence of function coefficients [67]. An appropriate set of Zernike functions were identified and the corresponding holograms were applied to correct the aberrations introduced by the optical system as well as the biological sample. Also, modal sensorless adaptive optics correction was reported for mapping optical aberration with 3D resolution in thick human skin biopsies and mouse brain tissues. These methods were integrated with a TPF microscope to analyse the consequences of tissue structure and refractive index distribution on aberration and imaging depth [65]. The intensity and resolution of TPF images were compared before and after aberration correction of GFP-stained cells in a fixed mouse embryo. The applied aberration correction for Fig. 5 is mostly significant in Zernike modes of astigmatism, coma and first-order spherical [65]. It was shown that aberration amplitude variations are strongly affected by the sample structure with imaging depth. In this analysis, the optimal correction was calculated over the entire acquired image.

Depth-resolved correction on a mouse embryo. The two images (left, before; right, after correction) share the same colour scale. Image size is ≈ 720 × 640 pixels. Insets, correction phase applied. b 3D reconstruction of a 30 × 40 × 40 µm3 region of the embryo. c Aberration amplitude in Zernike modes as a function of depth. The objective correction collar was set to optimize image quality at a depth of ≈ 50 µm in the sample. Figure is adapted with permission from [65]
Several studies have shown that to counteract the aberrations introduced by the sample, the exact wavefront shape has to be applied to the DMM to compensate the distorted wavefront of the excitation light [55]. The detection of aberrations in microscopy is straightforward. SH sensor is used to measure the wavefront of excitation light passes through a confocal pinhole [68]. This method uses the intensity or contrast of the fluorescence or backscattered light confined in a compact focal volume as a fitness factor which can be maximized using an optimization algorithm [69, 70]. A guide star was generated due to such a non-linear-excitation and used to improve the image quality of Caenorhabditis elegans in vivo [71]. Wang et al. demonstrated that high resolution and large volumes of neurons > 240 μm3 was achieved in zebrafish brains in vivo using a fluorescent guide star–based direct wavefront sensing in visible wavelengths [68]. The depth of imaging field was extended to 700 μm inside the mouse brain and resolve dendritic spines in vivo by near-infrared (NIR) wavelengths (see Fig. 6). Tissue scattering is reduced at longer wavelengths allowing direct wavefront sensing from fluorescent guide stars in the mouse brain [72]. The technique not only improves the visualization of morphological features of neurons but also their functional sensitivity at 500 μm depth in the primary visual cortex of the Thy1-GCaMP6s GP4.3 mice. The NIR fluorescence protein iRFP71316 (peak fluorescence emission at 713 nm) was used as a marker for TPF generated guide star down to 500 μm below pia to obtain corrective wavefronts. After AO correction, calcium detection sensitivity in the visual cortex was improved at depths of 400, 500 and 600 μm.

AO correction via direct wavefront sensing improves functional calcium imaging deep inside the cortex of a living mouse. a Calcium responses evoked by drifting-grating stimulation 400 and 500 μm below pia in the primary visual cortex of a mouse (Thy1-GCaMP6s line GP4.3) before (left panel) and after (right panel) correction. Brightness of each pixel reflects its s.d. across 800 frames imaged during five repetitions of the drifting-grating stimulus set and is correlated with the local calcium transient magnitude. Scale bar, 20 μm. b Calcium transients at regions of interest (ROI) i–vi, before and after AO correction. Solid colours label averaged transients; faded colours label transients during specific repetitions. Top panel indicates the orientations and drifting directions of the grating stimuli. Representative images from > 20 imaging sections in three mice. Figure is adapted with permission from [72]
For some applications where speed is not a limiting factor, due to large number of pixels and active area of SLM, it can be a good alternative to the wavefront sensor in terms of dynamic range and sensitivity. Daniel et al. implemented a pupil segmentation–based adaptive optical approach in a TPF microscope with full-pupil illumination [73]. In this case, a reflective liquid crystal phase–only SLM was used as the active optical element. The images were acquired by illuminating only a small part of the pupil at a time and wavefront aberration was reconstructed using the phase-shift values measured from the displacement of the corresponding excitation light in the focal plane. This approach recovers near diffraction-limited imaging performance and improves the imaging signal and resolution at a depth of 450 μm in a mouse cortex in vivo [73]. However, conventional adaptive optics systems are limited to a small imaging area but for large field of view multi-pupil adaptive optics (MPAO) enables wavefront correction (over 450 × 450 μm2). The approach was applied to high-speed 3D dynamic imaging of calcium, blood vasculature, microglia and structure of neuronal network in in vivo mammalian brain (see Fig. 7) [74].

Non-planar microscopy of 3D vasculature dynamics.a Vasculature network at 6–65 μm depth under the dura with the depth colour encoded. b Planar imaging of blood vessels at the depth of 35 μm. c Non-planar imaging of blood vessels and neural dendrites, with the relative depth shown in each image segment in microns. The central segment was at the depth of 35 μm. Magenta, Texas-Red-labelled blood plasma; green, YFP-expressing neural dendrites. d Wavefront applied to the pupil array, which shifted the focal planes and corrected tissue-induced aberration. Rad, radian. e Synchronized dilation of three blood vessels at different depths. Their corresponding positions are marked in c, with the dotted lines indicating the position of measurement. Vessel III is a vertically penetrating vessel originating from vessel II as shown by the volumetric view in a. Scale bars, 50 μm. The data shown in this figure are representative of three experiments. Figure is adapted with permission from [74]
Indirect (without wavefront sensor) and direct (wavefront sensor–based) methods differ in implementation, speed and sensitivity. The indirect sensing method is simpler in hardware implementation but slow in aberration measurement (seconds to minutes) and it is applicable to opaque tissue. Again, direct methods can find out corrective wavefront in weakly scattering samples within tens of milliseconds. Also, compare to iterative-based indirect methods, direct methods using guide stars may have higher accuracy in measuring the wavefront [75].
Summary and outlook
The recent advances in the field of neuroimaging have shed light on disease causing factors as well as brain circuit functioning. The use of confocal microscopy in brain imaging could now successfully provide information of upper layer neurons and the cellular signalling. However, to elucidate deeper regions of the brain tissue, TPF microscopy is widely used. This technique utilizes two-photon for excitation of the sample at focal point and provides information only from the focused region; hence, signal is free from out of focus area. Though TPF provides deep tissue imaging at sub-cellular level resolution, there remain limitations such as sample and optical aberrations. Until recently the invention of two separate techniques that use optical fibre and an endo-microscope supplemented with a GRIN lens, imaging deeper regions of the brain in a live animal was thought to be impossible. Computer-controlled phase–only SLM allows the dynamic control of the distribution of multiple excitation light for photo-stimulation of multiple regions of the sample. These multi-beam methods are currently limited in non-power limited setting applications but promise to be equally useful in power limited applications when more powerful lasers become available. The multiplex beam generation method is used extensively in neuronal circuits. However, the imaging depth is limited by the sample induced aberration.
The implementation of AO in TPF has revolutionized fluorescence imaging over the last decade [75,76,77] and in next few years, we will continue to witness unprecedented advances of this tool. Additional improvements in the developments will enhance our ability to image deeper into the brain and with better temporal resolution in vivo. Although AO is implemented efficiently in TPF microscope, spatial resolution remains limited by diffraction to ~ 0.3 μm laterally and ~ 0.8 μm axially. A multi-photon structured illumination microscopy is developed to determine optical aberrations and a deformable mirror to correct them uses a non-linear guide star. By combining two-photon and SLM diffraction-limited with deep penetration imaging, it is demonstrated on bead phantoms, cells in collagen gels, nematode larvae and embryos, drosophila brain and zebrafish embryos [78, 79].
Motivated by AO-based TPF for brain imaging, Bar-Noam and co-workers developed correction-free remotely scanned two-photon in vivo mouse retinal imaging [80]. This technique remotely scanned the laser beam and focus using an electronically tuneable lens that provides inertia free high-resolution 3D fluorescein angiograms at cellular resolution. Moreover, it allows long-term repeated imaging as well as functional calcium imaging of repeated retinal responses to light stimulation using the genetically encoded indicator, GCaMP6s. Despite the recent advances in TPF and adaptive optics for deep tissue imaging, neuroscientists and physicists are still trying to improve the existing techniques in order to achieve a much higher degree of cellular resolution that will eventually improve our understanding of brain structure and function. Application of AO for imaging deeper regions of the brain requires the identification of type of aberration and optimization of spatial and temporal resolutions to be achieved. Given the robustness of this method, it is crucial to identify the specimen-derived aberrations such as depth and transparency which can be corrected via AO. Direct or indirect methods of aberration corrections could be applied based on the specimen and laboratory needs. Once implemented, further improvements such as use of longer wavelength fluorescent probes can improve AO-based TPF for in vivo deep tissue imaging.
References
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C et al (2008) Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science 321:1686–1689
Miquelajauregui A, Kribakaran S, Mostany R, Badaloni A, Consalez GG, Portera-Cailliau C (2015) Layer 4 pyramidal neurons exhibit robust dendritic spine plasticity in vivo after input deprivation. J Neuroscience 35:7287–7294
Kim T, Oh WC, Choi JH, Kwon HB (2016) Emergence of functional subnetworks in layer 2/3 cortex induced by sequential spikes in vivo. Proc Natl Acad Sci U S A 113:E1372–E1381
Greenberg DS, Houweling AR, Kerr JN (2008) Population imaging of ongoing neuronal activity in the visual cortex of awake rats. Nature 11:749–751
Helmchen F, Denk W (2005) Deep tissue two-photon microscopy. Nat Methods 2:932–940
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318
Mertz J (2004) Nonlinear microscopy: new techniques and applications. Cur Opin Neurobiology 14:610–616
Meng G, Liang Y, Sarsfield S, Jiang WC, Lu R, Dudman JT et al (2019) High-throughput synapse-resolving two-photon fluorescence microendoscopy for deep-brain volumetric imaging in vivo. Elife 8:e40805
Piazza S, Bianchini P, Sheppard C, Diaspro A, Duocastella M (2018) Enhanced volumetric imaging in 2-photon microscopy via acoustic lens beam shaping. J Biophotonics 11:e201700050
Abdeladim L, Matho KS, Clavreul S, Mahou P, Sintes JM, Solinas X et al (2019) Multicolor multiscale brain imaging with chromatic multiphoton serial microscopy. Nat Commun 10:1662
Ricard C, Arroyo ED, He CX, Portera-Cailliau C, Lepousez G, Canepari M et al (2018) Two-photon probes for in vivo multicolor microscopy of the structure and signals of brain cell. Brain Struct Funct 223:3011–3043
Chen IW, Ronzitti E, Lee BR, Daigle TL, Dalkara D, Zeng H et al (2019) In vivo submillisecond two-photon optogenetics with temporally focused patterned light. J Neurosci 39:3484–3497
Ahn C, Hwang B, Nam K, Jin H, Woo T, Park JH (2019) Overcoming the penetration depth limit in optical microscopy: Adaptive optics and wavefront shaping. J. Innov. Opt. Health Sci 12:1930002
Rodríguez C, Ji N (2018) Adaptive optical microscopy for neurobiology. Cur Opin Neurobiol 50:83–91
Park JH, Yu Z, Lee K, Lai P, Park Y (2018) Perspective: wavefront shaping techniques for controlling multiple light scattering in biological tissues: Toward in vivo applications. APL Photonics 3:100901
Turcotte R, Liang Y, Ji N (2017) Adaptive optical versus spherical aberration corrections for in vivo brain imaging. Biomed Opt Express 8:3891–3902
Bueno JM, Skorsetz M, Bonora S, Artal P (2018) Wavefront correction in two-photon microscopy with a multi-actuator adaptive lens. Opt Express 26:14278–14287
Galwaduge P, Kim S, Grosberg L, Hillman E (2015) Simple wavefront correction framework for two-photon microscopy of in-vivo brain. Biomed Opt Express 6:2997–3013
Wilt BA, Burns LD, Wei Ho ET, Ghosh KK, Mukamel EA, Schnitzer MJ (2009) Advances in light microscopy for neuroscience. Ann Rev Neurosci 3:435–506
Matsumoto N, Inoue T, Matsumoto A, Okazaki S (2015) Correction of depth-induced spherical aberration for deep observation using two-photon excitation fluorescence microscopy with spatial light modulator. Biomed Opt Express 6:2575–2587
Mittmann W, Wallace DJ, Czubayko U, Herb JT, Schaefer AT, Looger LLO et al (2011) Two-photon calcium imaging of evoked activity from L5 somatosensory neurons in vivo. Nat Neurosci 14:1089–1093
Grinvald A, Frostig RD, Siegel RM, Bartfeld E (1991) High-resolution optical imaging of functional brain architecture in the awake monkey. Proc Natl Acad Sci U S A 88:11559–11563
Lu L, Gutruf P, Xia L, Bhatti LD, Wang X et al (2018) Wireless optoelectronic photometers for monitoring neuronal dynamics in the deep brain. Proc Natl Acad Sci U S A 115:E1374–E138s3
Denk W, Strickler JH, Webb WW (1990) Two-photon laser scanning fluorescence microscopy. Science 248:73
Alvarez VA, Sabatini BL (2007) Anatomical and physiological plasticity of dendritic spines. Ann Rev Neurosci 30:79–97
Yaseen MA, Sakadžić S, Wu W, Becker W, Kasischke KA, Boas DA (2013) In vivo imaging of cerebral energy metabolism with two-photon fluorescence lifetime microscopy of NADH. Biomed Opt Express 4:307–321
Wang HK, Majewska A, Schummers J, Farley B, Hu C, Sur M, Tonegawa S (2006) In vivo two-photon imaging reveals a role of arc in enhancing orientation specificity in visual cortex. Cell 126:389–402
Birkner A, Tischbirek CH, Konnerth A (2017) Improved deep two-photon calcium imaging in vivo. Cell Calcium 64:29–35
Helmchen F, Svoboda K, Denk W, Tank DW (1999) In vivo dendritic calcium dynamics in deep-layer cortical pyramidal neurons. Nat Neurosci 2:11
Stosiek C, Garaschuk O, Holthoff K, Konnerth A (2003) In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A 100:7319–7324
Theer P, Denk W (2006) On the fundamental imaging-depth limit in two-photon microscopy. JOSA A 23:3139–3149
Yang W, Carrillo-Reid L, Bando Y, Peterka DS, Yuste R (2018) Simultaneous two-photon imaging and two-photon optogenetics of cortical circuits in three dimensions. Elife 7:e32671
Kawakami R., Sawada K., Sato A., Hibi T., Kozawa Y., Sato S., et al. (2013) Visualizing hippocampal neurons with in vivo two-photon microscopy using a 1030 nm picosecond pulse laser, Sci Rep, 3.
Yasuda R, Harvey CD, Zhong H, Sobczyk A, Van Aelst L, Svoboda K (2006) Supersensitive Ras activation in dendrites and spines revealed by two-photon fluorescence lifetime imaging. Nat Neurosci 9:283
Zong W, Wu R, Li M, Hu Y, Li Y, Li J et al (2017) Fast high-resolution miniature two-photon microscopy for brain imaging in freely behaving mice. Nat Methods 14:713
Peters AJ, Liu H, Komiyama T (2017) Learning in the rodent motor cortex. Ann Rev Neurosci 40:77–97
Ziv Y, Burns LD, Cocker ED, Hamel EO, Ghosh KK, Kitch LJ et al (2013) Long-term dynamics of CA1 hippocampal place codes. Nat Neurosci 16:264–266
Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM et al (2013) Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494:238
Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A et al (2014) Natural neural projection dynamics underlying social behaviour. Cell 157:1535–1551
Resendez SL, Stuber GD (2015) In vivo calcium imaging to illuminate neurocircuit activity dynamics underlying naturalistic behaviour. Neuropsychopharmacology 40:238
Barretto RP, Ko TH, Jung JC, Wang TJ, Capps G, Waters AC et al (2011) Time-lapse imaging of disease progression in deep brain areas using fluorescence microendoscopy. Nat Med 17:223–228
Delgado E. M., Psaltis D., Moser C. (2016) Two-photon excitation endoscopy through a multimode optical fiber, in: SPIE BiOS, International Society for Optics and Photonics, 97171E–97171E.
Bocarsly ME, Jiang WC, Wang C, Dudman JT, Ji N, Aponte Y (2015) Minimally invasive microendoscopy system for in vivo functional imaging of deep nuclei in the mouse brain. Biomed Opt Express 6:4546–4556
Champelovier D, Teixeira J, Conan JM, Balla N, Mugnier LM, Tressard T et al (2017) Image-based adaptive optics for in vivo imaging in the hippocampus. Sci Rep 7:42924
Forli A, Vecchia D, Binini N, Succol F, Bovetti S, Moretti C et al (2018) Two-photon bidirectional control and imaging of neuronal excitability with high spatial resolution in vivo. Cell Rep 22:3087–3098
Kramer RH, Fortin DL, Trauner D (2009) New photochemical tools for controlling neuronal activity. Curr Opin Neurobiol 19:544–552
Bednarkiewicz A, Bouhifd M, Whelan MP (2008) Digital micromirror device as a spatial illuminator for fluorescence lifetime and hyperspectral imaging. Appl Opt 47:1193–1199
Gustafsson MG, Shao L, Carlton PM, Wang CR, Golubovskaya IN, Cande WZ et al (2008) Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophy J 94:4957–4970
Losavio BE, Iyer V, Saggau P (2009) Two-photon microscope for multisite microphotolysis of caged neurotransmitters in acute brain slices. J Biomed Opt 14:064033–064014
Dal MM, Difato F, Beltramo R, Blau A, Benfenati F, Fellin T (2010) Simultaneous two-photon imaging and photo-stimulation with structured light illumination. Opt Express 18:18720–18731
Lutz C, Otis TS, DeSars V, Charpak S, DiGregorio DA, Emiliani V (2008) Holographic photolysis of caged neurotransmitters. Nat Methods 5:821–827
Feeks JA, Hunter JJ (2017) Adaptive optics two-photon excited fluorescence lifetime imaging ophthalmoscopy of exogenous fluorophores in mice. Biomed Opt Express 8:2483–2495
Mostany R., Miquelajauregui A., Shtrahman M., Portera-Cailliau C. (2015) Two-photon excitation microscopy and its applications in neuroscience, Advanced fluorescence microscopy: Methods and Protocols, 25-42.
Gould TJ, Burke D, Bewersdorf J, Booth MJ (2012) Adaptive optics enables 3D STED microscopy in aberrating specimens. Opt Express 20:20998–21009
Facomprez A, Beaurepaire E, Débarre D (2012) Accuracy of correction in modal sensorless adaptive optics. Opt Express 20:2598–2612
Packer MA, Russell EL, Dalgleish WPH, Häusser M (2015) Simultaneous all-optical manipulation and recording of neural circuit activity with cellular resolution in vivo. Nat Methods 12:140–146
Nikolenko V., Watson B. O., Araya R., Woodruff A., Peterka D. S., Yuste R. (2008) SLM microscopy: scanless two-photon imaging and photostimulation with spatial light modulators, Front Neural Circuits, 2.
Wang K, Milkie DE, Saxena A, Engerer P, Misgeld T, Bronner ME et al (2014) Rapid adaptive optical recovery of optimal resolution over large volumes. Nat Methods 11:625–628
Tao X, Norton A, Kissel M, Azucena O, Kubby J (2013) Adaptive optical two-photon microscopy using autofluorescent guide stars. Opt Lett 38:5075–5078
Quirin S, Jackson J, Peterka DS, Yuste R (2014) Simultaneous imaging of neural activity in three dimensions. Front Neural Circuits 8:29
Yang W, Miller JE, Carrillo-Reid L, Pnevmatikakis E, Paninski L, Yuste R et al (2016) Simultaneous multi-plane imaging of neural circuits. Neuron 89:269–284
Champelovier D., Teixeira J., Conan J. M., Balla N., Mugnier L., Tressard T., et al (2017) Image-based adaptive optics for in vivo imaging in the hippocampus, Sci Rep, 7.
Skorsetz M, Artal P, Bueno JM (2016) Performance evaluation of a sensorless adaptive optics multiphoton microscope. J Microscopy 261:249–258
Marsh P, Marsh D, Girkin J (2003) Practical implementation of adaptive optics in multiphoton microscopy. Opt Express 11:1123–1130
Débarre D, Botcherby EJ, Watanabe T, Srinivas S, Booth MJ, Wilson T (2009) Image-based adaptive optics for two-photon microscopy. Opt Lett 34:2495–2497
Zeng J, Mahou P, Schanne-Klein MC, Beaurepaire E, Débarre D (2012) 3D resolved mapping of optical aberrations in thick tissues. Biomed Opt Express 3(1898-1913):2012
Booth MJ (2014) Adaptive optical microscopy: the ongoing quest for a perfect image. Light Sci Appl 3:e165
Cha JW, Ballesta J, So PT (2010) Shack-Hartmann wavefront-sensor-based adaptive optics system for multiphoton microscopy. J Biomed Optics 15:046022–046010
Rueckel M, Mack-Bucher JA, Denk W (2006) Adaptive wavefront correction in two-photon microscopy using coherence-gated wavefront sensing. Proc Natl Acad Sci U S A 103:17137–17142
Ji N, Sato TR, Betzig E (2012) Characterization and adaptive optical correction of aberrations during in vivo imaging in the mouse cortex. Proc Natl Acad Sci U S A 109:22–27
Aviles-Espinosa R, Andilla J, Porcar-Guezenec R, Olarte OE, Nieto M, Levecq X et al (2011) Measurement and correction of in vivo sample aberrations employing a nonlinear guide-star in two-photon excited fluorescence microscopy. Biomed Opt Express 2:3135–3149
Wang K., Sun W., Richie C. T., Harvey B. K., Betzig E., Ji N. (2015) Direct wavefront sensing for high-resolution in vivo imaging in scattering tissue, Nat Commun, 6.
Ji N, Milkie DE, Betzig E (2010) Adaptive optics via pupil segmentation for high-resolution imaging in biological tissues. Nat Methods 7:141–147
Park JH, Kong L, Zhou Y, Cui M (2017) Large-field-of-view imaging by multi-pupil adaptive optics. Nat Methods 14:581–583
Ji N (2017) Adaptive optical fluorescence microscopy. Nat Methods 14:374–380
Yang W, Yuste R (2017) In vivo imaging of neural activity. Nat Methods 14:349–359
Gautam V, Drury J, Choy JM, Stricker C, Bachor HA, Daria VR (2015) Improved two-photon imaging of living neurons in brain tissue through temporal gating. Biomed Opt Express 6:4027–4036
Winter PW, York AG, Dalle ND, Ingaramo M, Christensen R, Chitnis A et al (2014) Two-photon instant structured illumination microscopy improves the depth penetration of super-resolution imaging in thick scattering samples. Optica 1:181–191
Zheng W, Wu Y, Winter P, Fischer R, Nogare DD, Hong A et al (2017) Adaptive optics improves multiphoton super-resolution imaging. Nat Methods 14:869
Bar-Noam AS, Farah N, Shoham S (2016) Correction-free remotely scanned two-photon in vivo mouse retinal imaging. Light Sci Appl 5:e16007
Acknowledgements
We thank Dr. K. Satyamoorthy, Director, Manipal School of Life Sciences (MSLS), Manipal for his encouragement. Authors thank Dr. K. K. Mahato, Head of Department of Biophysics, MSLS for his constant support and Manipal Academy of Higher Education (MAHE), Manipal, India, for providing the infrastructure needed.
Funding
We reeived financial support from the SERB-Department of Science and Technology (DST), Government of India (Project Number—ECR/2016/001944).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sahu, P., Mazumder, N. Advances in adaptive optics–based two-photon fluorescence microscopy for brain imaging. Lasers Med Sci 35, 317–328 (2020). https://doi.org/10.1007/s10103-019-02908-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-019-02908-z