
Abstract
Mesenchymal stem cells (MSCs) are promising for use in regenerative medicine. Low-level light irradiation (LLLI) has been shown to modulate various processes in different biological systems. The aim of our study was to investigate the effect of red light emitted from a light-emitting diode (LED) on bone marrow MSCs with or without osteogenic supplements. MSCs both with and without osteogenic supplements were divided into four groups, and each group was irradiated at doses of 0, 1, 2 and 4 J/cm2. Cellular proliferation was evaluated using WST-8 and 5-ethynyl-2′-deoxyuridine (EdU) fluorescence staining. The alkaline phosphatase activity, mineralization, and expression of osteoblast master genes (Col1α1, Alpl, Bglap and Runx2) were monitored as indicators of MSC differentiation towards osteoblasts. In groups without osteogenic supplements, red light at all doses significantly stimulated cellular proliferation, whereas the osteogenic phenotype of the MSCs was not enhanced. In groups with osteogenic supplements, red light increased alkaline phosphatase activity and mineralized nodule formation, and stimulated the expression of Bglap and Runx2, but decreased cellular proliferation. In conclusion, nonconherent red light can promote proliferation but cannot induce osteogenic differentiation of MSCs in normal media, while it enhances osteogenic differentiation and decreases proliferation of MSCs in media with osteogenic supplements.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Non-union and delayed healing are major complications in orthopedic patients with bone fractures. As a novel reconstructive therapy, bone marrow mesenchymal stem cells (MSCs) are receiving attention because of their easy isolation, expansion, low immunogenicity and potential for long-term generation, making them ideal for repairing bone defects in bone bioengineering. Bone MSCs are derived from bone marrow. They exhibit a fibroblast-like appearance, and can differentiate in vitro into various anchorage-dependent cell types including but not limited to bone, cartilage and adipose tissue [1–5]. Differentiation of MSCs can be induced by the local extracellular matrix [6], growth factors [7] and some physical factors [8].
Low-level light irradiation (LLLI) has been shown to modulate various processes in different biological systems, for example promoting wound healing [9, 10], cellular proliferation [11, 12], collagen synthesis [13] and cytokine production [14, 15]. The absorption of low-intensity laser light by biological systems is of a purely noncoherent nature. The coherent properties of laser light are not manifested at the molecular level [16]. It is considered that laser and LED light produce similar biological responses at the cellular level. The effect of photoinduced osteodifferentiation is still controversial, as previous studies have shown different and conflicting results. Oliveira et al. [17] used a diode laser (830 nm, 2.86 J/cm2) to irradiate odontoblast-like cells. Neither the metabolism (as evaluated using the MTT assay) nor mRNA expression of collagen in the irradiated cells differed significantly from those in the nonirradiated cells. Irradiated cells also showed lower alkaline phosphatase (ALP) activity than the nonirradiated cells. In contrast, Ozawa et al. [18] reported that laser irradiation (830 nm, 3.82 J/cm2) at an earlier stage of bone formation is more effective than irradiation at a later stage, and that stimulation of bone formation by laser is dependent on the total energy dose. In a recent study, Abramovitch-Gottlib et al. [19] found that a low-energy laser stimulated the osteogenic phenotype of MSCs in a three-dimensional biomatrix. Kim et al. [20] also combined 647 nm red light and osteogenic differentiation media (ODM) to induce differentiation of MSCs towards osteoblasts. However, the effects of LLLI on osteodifferentiation have not yet been elucidated.
In this work, we investigated the effects of red light emitted from an LED on bone marrow MSCs cultured in either normal medium or ODM.
Materials and methods
MSC isolation and culture
The procedures regarding osteogenic cell isolation and culture have been described by Jiang et al. [21]. Briefly, a 4-week-old Sprague-Dawley rat was killed by neck dislocation according to the Guidelines of the Animal Care and Use Committee for Teaching and Research of Huazhong University of Science and Technology. Bone marrow was collected from the femurs and tibiae with a needle, suspended in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Grand Island, NY), and centrifuged at 2,000 rpm for 5 min. The marrow pellet was washed in phosphate-buffered saline, centrifuged at 1,000 rpm for 10 min, and then resuspended in DMEM. Nucleated cells were isolated with a Percoll density gradient (Invitrogen) by centrifuging at 14,000 rpm for 12 min. The top 60% of the gradient was collected, and then washed with the complete culture medium containing 10% fetal bovine serum (Invitrogen), 100 U/ml penicillin (Sigma-Aldrich, St Louis, MO), 100 mg/ml streptomycin (Sigma-Aldrich), and 0.25 mg/ml amphotericin (Sigma-Aldrich). The cells were then seeded in a tissue culture flask and incubated in the medium as described above with 10 ng/ml leukemia inhibitory factor (Invitrogen) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Nonadherent cells were removed by changing the medium after 24 h. The culture medium was changed twice a week thereafter. For subculture, cells were detached with 0.25% trypsin (Amresco, Solon, OH) and passaged at a ratio of 1:2 when they had grown to 80–90% confluence. The cells were plated onto 35-mm tissue-culture dishes (Greiner, Frickenhausen, Germany) at a density of 8 × 105 cells/dish, and 96-well ELISA plates (Jet-Biofil, Guangzhou, China) at a density of 3 × 103 cells/well. After incubation for 24 h, the medium in half the dishes/wells was changed to ODM (Cyagen Biosciences, Guangzhou, China) which consisted of low-glucose DMEM supplemented with 50 μg/ml ascorbic acid, 10–8 M dexamethasone, and 10 mM β-glycerolphosphate. The medium in the remaining half was not changed from DMEM.
Irradiation procedure
A red-light LED (LL-HP60VC; Jiaguang, Beijing, China) with a continuous output and a wavelength of 620 nm (1 W output) was used in this study. The distance from the LED to the cell layer was 2 cm. The diameter of the light spot was 10 cm. At the cell-layer level, the power density measured using a power meter (Ophir Optronics, Jerusalem, Israel) was 6.67 mW/cm2. Because biostimulation after irradiation may continue for 48 h [22], the cultures were irradiated every other day since in half of them the medium had been changed to ODM. The first irradiation day was set as day 0. The duration of each irradiation for red light was calculated as 150 s for 1 J/cm2, 300 s for 2 J/cm2 and 600 s for 4 J/cm2. Two cultures were used as controls: MSCs incubated in DMEM without irradiation (control 1), and MSCs incubated in ODM without irradiation (control 2). Nonirradiated cells were maintained outside the incubator under the same conditions as the irradiated cells.
Cell proliferation assays
Since the distribution of the light energy density from a LED is not uniform, the energy dosages absorbed by different wells in 96-well microplates are significantly different [23]. We seeded cells in disassembled wells of ELISA plates, and every well was placed at the same point and irradiated separately. Cell viability was assessed with a WST-8 kit (Beyotime Institute of Biotechnology, Jiangsu, China) at 2, 4, 6 and 8 days, respectively. At the indicated times, WST-8 was added to the cells, according to the manufacturer’s instructions, followed by incubation for 1 h. OD450, the absorbance value at a wavelength of 450 nm, was read in an ELX 800 universal microplate reader (Bio-Tek Instruments, Winooski, VT). The value is directly proportional to the number of viable cells in a culture medium and cell proliferation.
According to the results of WST-8 assay, we also investigated the newly synthesized DNA of replicating cells irradiated with 2 J/cm2 on day 4 using a combination of Hoechst 33342 staining and a Cell-Light EdU DNA cell proliferation kit [24] according to the manufacturer’s instructions (Guangzhou RiboBio, Guangzhou, China). EdU-labeled cells were counted manually in ten fields of view randomly selected from each well, and percentages were calculated.
Alkaline phosphatase activity assay
ALP activity peaks at the end of the proliferative stage and before matrix maturation [25]. According to some studies [8, 26], osteogenically induced cells would show a peak of ALP activity approximately between 7 days and 14 days after they had been seeded in the osteogenic differentiation system. An ALP activity assay was performed at 3, 7 and 10 days after the first irradiation. The cells were rinsed twice with phosphate-buffered saline, then 0.1% Triton X-100 containing 10 mmol/l Tris-HCl, 5 mmol/l MgSO4 and 0.1% NaNO3 was added to 500 μl, and the cells were frozen at −80°C three times to disrupt the cell membranes. ALP activity and protein content were measured using an ALP activity kit (Nanjing Jiancheng Biological Engineering Institute, Nanjing, China) and a Micro BCA assay kit (Lipulai, Beijing, China). All results were normalized in relation to protein content [27].
Mineralization assay
For mineralization assay, calcium phosphate hydroxyapatite was detected using a commercial von Kossa staining kit (Genmed Scientifics, Shanghai, China) on day 21. The mineral deposit area of each dish was calculated from the positively stained areas measured in 20 randomly selected microscopic fields (96,467.83 μm2/field) using Image-Pro1 Plus analysis software (Media Cybernetics, Silver Spring, MD) [18].
RNA preparation and RT-PCR analysis
When cells had reached approximately 90% confluence, the medium of half the dishes was changed to ODM. After 3 days of incubation in ODM, the cells were treated with red light. Total RNA of bone marrow MSCs cultured in 35-mm dishes was extracted after irradiation by an acid guanidinium thiocyanate/phenol/chloroform extraction method. Cells were homogenized using TRIZOL reagent (Invitrogen), and total RNA was isolated. RNA was reverse-transcribed (RT) with a reverse transcriptase kit (Toyoba, Osaka, Japan). According to the manufacturer’s instructions, RT was performed in a 20-μl reaction mixture. Aliquots of cDNA were amplified in a 25-μl polymerase chain reaction (PCR) reaction mixture which contained 10 nM 5′- and 3′-oligomers. The expression levels of collagen type I (Col1α1, an early marker of osteodifferentiation in proliferative stage), ALP (Alpl, a medium-term marker of osteodifferentiation before matrix maturation), osteocalcin (Bglap, a late period marker in matrix maturation), and Runx2 (also called Cbfa1, initially expressed in osteoprogenitor cells) [28, 29] were determined. We designed the PCR primers with Primer 3 (http://frodo.wi.mit.edu/primer3/). The gene sequences were searched in MEDLINE. The primer sequences are shown in Table 1.
Statistical analysis
All data are expressed as means ± standard deviation (n = 3). The analyses were performed with SPSS 13.0 software (SPSS, Chicago, IL). Statistical analysis was done using a one-way ANOVA. All comparisons were evaluated at a 95% confidence level (P < 0.05).
Results
Effect of LLLI on cellular proliferation
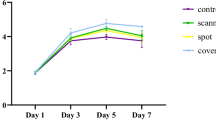
As shown in Fig. 1, viable cell numbers increased rapidly from day 0 (24 h after cell seeding) to day 4, and then reached a stationary phase by day 6. Similar cell growth curves were observed in every group throughout the cell culture period. At 4, 6 and 8 days, cells cultured in DMEM showed significantly higher viabilities than that cells cultured in ODM. Among the DMEM cultures, red light at all doses significantly stimulated cell viability as compared with the control 1 culture. Cultures irradiated at 1 J/cm2 had the fastest proliferation rates. Their OD450 was significantly higher than that of the control 1 culture on day 4 (P = 0.0034, <0.01) and day 6 (P = 0.017, <0.05). Cultures irradiated at 2 J/cm2 also showed a higher OD450 on day 4 (P = 0.0067, <0.01) and day 6 (P = 0.023, <0.05) than the control 1 culture. Cultures irradiated at 4 J/cm2 only showed a significantly higher OD450 on day 4 (P = 0.039, <0.05). Among the ODM cultures, the control 2 culture and those irradiated at 1 J/cm2 showed similar proliferation rates. Red light at 2 and 4 J/cm2 significantly inhibited cell viability as compared with the control 2 culture. Irradiation at 2 J/cm2 resulted in a lower OD450 on day 6 (P = 0.036, <0.05) compared with the control 2 culture. The slowest proliferation rate was induced by irradiation at 4 J/cm2, as lower OD450 values were observed on day 4 (P = 0.0057, <0.01) and day 6 (P = 0.028, <0.05).

Effect of LLLI on viability of rat bone marrow MSCs. Irradiated MSCs in DMEM showed a significant increase in viability on days 4 and 6 as compared to those in the control 1 culture. The final saturation densities were not significantly different among the DMEM cultures, whereas they were significantly higher than in the control 2 culture. The final saturation densities were not significantly different among the ODM cultures. On day 4, cell viability following LLLI at 4 J/cm2 was significantly lower than in the control 2 culture. On day 6, cell viability following LLLI at 2 and 4 J/cm2 was significantly lower than in the control 2 culture (**P<0.01, *P<0.05 vs. control 1; ## P<0.01, # P<0.05 vs. control 2; n = 3)
EdU labeling was performed on day 4. As shown in Fig. 2, In the DMEM cultures, more irradiated cells showed red fluorescence indicating EdU labeling (P = 0.029, <0.05). Among the ODM cultures, there were fewer EdU-labeled cells in the control 2 culture (P = 0.021, <0.05) and irradiated cultures (P = 0.013, <0.05) than in the control 1 culture. However, red light irradiation did not induce significant differences among the ODM cultures.

Effect of LLLI on DNA synthesis in rat bone marrow MSCs. The experiment was performed on day 4. All cell nuclei showed blue fluorescence indicating Hoechst 33342 staining, and EdU labeling showed replicating cells. a Among the DMEM cultures, more cells showed red fluorescence indicating EdU labeling following LLLI. b Among the ODM cultures, LLLI had no significant effect. c Analysis of EdU-labeled cells. Among the DMEM cultures, more irradiated cells showed red fluorescence indicating EdU labeling. Among the ODM cultures, there were significantly fewer EdU-labeled cells in the control 2 culture and irradiated cultures than in the control 1 culture. However, red light did not induce significant difference among the ODM cultures (*P<0.05 vs. control 1; # P<0.05 vs. control 1)
Effect of LLLI on cellular differentiation
As shown in Fig. 3, all DMEM cultures showed similar ALP activity levels which were clearly lower than that in the ODM cultures. Their ALP activities did not significantly fluctuate with the changes in incubation time and irradiation dose. Among the ODM cultures, irradiation at 4 J/cm2 significantly increased ALP activity as compared with the control 2 culture on day 3 (1.26-fold, P = 0.034) and day 7 (1.32-fold, P = 0.021). ALP activities of MSCs in ODM reached a peak on day 10, and cultures irradiated at 2 J/cm2 showed significantly higher ALP activity than the control 2 culture (1.22-fold, P = 0.032).

Effect of LLLI on ALP activity in rat bone marrow MSCs. Analysis was performed on days 7, 10 and 14 after the first irradiation. The data presented are means ± standard deviation (n = 3) (# P<0.05 vs. control 1; *P<0.05 vs. control 2)
A well-delineated, three-dimensional nodular structure of mineralized tissue was formed by MSCs cultured in ODM, while no nodular structure was found in MSCs cultured in DMEM (Fig. 4). The number of bone nodules present in 35-mm dishes with ODM was counted, and significant stimulation by red light irradiation occurred at all energy doses. The staining density in dishes was significantly stimulated by LLLI. The control 2 culture was considered as standard (100%). MSCs in ODM irradiated at doses of 2 and 4 J/cm2 showed significant increases in staining intensity of 134% (P = 0.024) and 166% (P = 0.013), respectively.

Effect of LLLI on mineral nodule formation in rat bone marrow MSCs. a Von Kossa staining of MSCs on day 21. Mineral nodules formed in ODM cultures, whereas no mineral nodules formed in DMEM cultures. b Analysis of mineral nodule formation in ODM cultures. MSCs in ODM irradiated at doses of 2 and 4 J/cm2 showed increases in staining intensity of 134% and 166%, respectively, versus control 2 (*P<0.05 vs. control 2)
As shown in Fig. 5, the mRNA expression levels of Col1α1, Alp1, Bglap and Runx2 in the irradiated cells were compared with those of cells in the control 1 and control 2 cultures. In the DMEM cultures, none of the genes showed significant differences in mRNA expression, but the mRNA expression levels of irradiated cells were clearly lower than that in cells of the control 2 culture. In ODM cultures, LLLI had no significant effect on the expression levels of Col1α1 and Alp1, but significantly increased the levels of Bglap and Runx2. The expression levels of Bglap mRNA following irradiation at doses of 1, 2 and 4 J/cm2 were 97% (P = 0.27), 153% (P = 0.026) and 143% (P = 0.037), respectively, and the levels of Runx2 were 126% (P = 0.029), 188% (P = 0.014) and 129% (P = 0.032), respectively, versus the expression level in the control 2 culture.

The mRNA expression levels of Col1α1, Alp1, Bglap and Runx2. Total RNA was isolated on day 4 (*P<0.05 vs. control 2)
Discussion
In the present study, the effects of 620-nm red light at different energy dosages on bone marrow MSCs were investigated. The major findings were: (1) red light promoted proliferation of MSCs cultured in normal medium, and suppressed proliferation of MSCs cultured in ODM; (2) red light enhanced osteogenic differentiation of MSCs cultured in ODM, but did not induce osteogenic differentiation of MSCs cultured in normal medium.
Some studies have shown that proliferation of MSCs can be promoted by laser irradiation [11, 12, 30, 31]. Karu [16] and Posten et al. [32] suggested that coherent and noncoherent light with the same wavelength, intensity and irradiation time provide similar effects. It seems reasonable to assume that LED light would have the ability to affect proliferation of MSCs. In our study, the results of the WST-8 and EdU assays confirmed that noncoherent light is also able to stimulate proliferation of bone marrow MSCs cultured in normal medium. However, irradiation with red light slowed down cellular proliferation of MSCs cultured in ODM. This reduced rate of proliferation was matched by an increased rate of osteodifferentiation of MSCs in ODM. A possible explanation is that there is a reciprocal relationship between growth and osteogenic differentiation in MSCs [28]. Genes involved in the production and deposition of the extracellular matrix are expressed during the proliferative period, and the synthesis of an organized bone-specific extracellular matrix contributes to the shutdown of proliferation [33]. This reciprocal relationship also suggests that the induction of osteogenic differentiation by red light irradiation occurs during the proliferative stages. Moreover, cultures of irradiated cells showed final saturation densities similar to those in the corresponding ODM control culture, whereas irradiated cultures showed a clear enhancement of the intensity of von Kossa staining compared with that of the control 2 culture. Therefore, it seems that the significant increase in the number of nodules following red light irradiation was not attributable to a general increase in cell number, but rather to an increase in the percentage of cells showing mineralization.
MSCs are pluripotent and can differentiate into multiple cell lineages [1–5], but they do not differentiate spontaneously. Appropriate in vitro conditions is a necessary factor for osteodifferentiation. In this study, 620-nm red LED light significantly enhanced ALP activity and mineral deposition which correlated with osteodifferentiation of MSCs cultured in ODM. As shown here, red light enhanced the expression of Runx2 and Bglap, which are osteogenesis markers of osteoblast differentiation [34, 35]. Since Runx2 and Osteocalcin can both be expressed at the same time before the mature osteoblast phase [29, 36], the RT-PCR results indicated that the primary induction of osteogenic differentiation by red light irradiation appeared to be commitment to the osteoblast pathway through an early increase in Runx2 gene expression, rather than at the level of osteoblast maturation. On the other hand, although the MSCs in DMEM were also irradiated at the same doses, they did not express osteogenic phenotypes, such as ALP activity, mineral matrix formation and upregulation of osteoblast master gene. We speculate that red light alone cannot activate the signaling pathway that controls osteodifferentiation. However, it could intervene in the differentiation signaling pathway by some means still unknown if the signaling pathway had already been activated. In other words, red light irradiation could be an adjunctive tool to increase osteodifferentiation, although it cannot induce osteodifferentiation alone.
It is now well established that osteodifferentiation is marked by sequential stages of cellular proliferation and bone extracellular matrix maturation [25]. ALP activity is a transient early marker of osteodifferentiation in MSCs known to peak at the end of the proliferative stage and before matrix maturation [33]. Early progenitor cells do not express ALP activity, but differentiate through a defined number of cell divisions, ultimately expressing a mature osteoblast phenotype: a postmitotic, osteogenic cell with ALP activity [37]. In the ODM cultures in our experiments, the ALP mRNA expression in all red light irradiated cultures did not show significant difference from that in the control 2 culture, whereas the cultures irradiated at 2 J/cm2 and 4 J/cm2 showed a significantly higher ALP activity than others. ALP is synthesized as an inactive precursor, pro-ALP, and is processed into an active form by proteolytic cleavage [38]. The ALP mRNA expression corresponds to the amount of pro-ALP, whereas hydrolyzed p-nitrophenyl phosphate reflects the amount of activated ALP. Red light irradiation could enhance the proteolytic cleavage of pro-ALP.
To sum up, we investigated the role of noncoherent red light irradiation in photoinduced osteogenic differentiation by determining the cellular proliferation and osteodifferentiation effects of LED light on bone marrow MSCs cultured in two different biological systems. Irradiated MSCs in two different in vitro environments showed different behaviors. Red LED light at different energy densities promoted proliferation but did not induce osteogenic differentiation of MSCs in normal medium, while it enhanced osteogenic differentiation and decreased the rate of proliferation of MSCs in ODM. We conclude that red light can effectively increase the rate of osteodifferentiation despite its inability to induce osteodifferentiation alone. This result will help us to understand the mechanisms of action of light on bone. Our findings may contribute to regenerative medical treatment of bone fractures, since they provide the basis for a cheap and efficient strategy for controlling the expansion and differentiation of MSCs in vitro prior to transplantation.
Reference
Prockop DJ (1997) Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 276:71–74
Caplan AI, Bruder S (2001) Mesenchymal stem cells: building blocks for molecular medicine in the 21st Century. Trends Mol Med 7:259–264
Dennis JE, Charbord P (2002) Origin and differentiation of human and murine stroma. Stem Cells 20:205–214
Tondreau T, Lagneaux L, Dejeneffe M, Massy M, Mortier C, Delforge A, Bron D (2004) Bone marrow-derived mesenchymal stem cells already express specific neural proteins before any differentiation. Differentiation 72:319–326
Caplan AI (2005) Review: mesenchymal stem cells: cell-based reconstructive therapy in orthopedics. Tissue Eng 11:1198–1211
Engler AJ, Sen S, Sweeney HL, Discher DE (2006) Matrix elasticity directs stem cell lineage specification. Cell 126:677–689
Liu ZJ, Zhuge Y, Velazquez OC (2009) Trafficking and differentiation of mesenchymal stem cells. J Cell Biochem 106:984–991
Green DE, Longtin JP, Sitharaman B (2009) The effect of nanoparticle-enhanced photoacoustic stimulation on multipotent marrow stromal cells. ACS Nano 3:2065–2072
Mester E, Nagylucskay S, Tisza S, Mester A (1978) Stimulation of wound healing by means of laser rays. Part III – Investigation of the effect on immune competent cells. Acta Chir Acad Sci Hung 19:163–170
Mester E, Mester AF, Mester A (1985) The biomedical effects of laser application. Lasers Surg Med 5:31–39
Tuby H, Maltz L, Oron U (2007) Low-level laser irradiation (LLLI) promotes proliferation of mesenchymal and cardiac stem cells in culture. Lasers Surg Med 39:373–378
Hou JF, Zhang H, Yuan X, Li J, Wei YJ, Hu SS (2008) In vitro effects of low-level laser irradiation for bone marrow mesenchymal stem cells: proliferation, growth factors secretion and myogenic differentiation. Lasers Surg Med 40:726–733
Pereira AN, Eduardo Cde P, Matson E, Marques MM (2002) Effect of low-power laser irradiation on cell growth and procollagen synthesis of cultured fibroblasts. Lasers Surg Med 31:263–267
Kipshidze N, Nikolaychik V, Keelan MH, Shankar LR, Khanna A, Kornowski R, Leon M, Moses J (2001) Low-power helium:neon laser irradiation enhances production of vascular endothelial growth factor and promotes growth of endothelial cells in vitro. Lasers Surg Med 28:355–364
Byrnes KR, Wu X, Waynant RW, Ilev IK, Anders JJ (2005) Low power laser irradiation alters gene expression of olfactory ensheathing cells in vitro. Lasers Surg Med 37:161–171
Karu TI (2003) Low-power laser therapy. In: VoDinh T (ed) Biomedical photonics handbook. CRC Press, Boca Raton, pp 1–25
Oliveira CF, Hebling J, Souza PPC, Sacono NT, Lessa FR, Lizarelli RFZ, Costa CAS (2008) Effect of low-level laser irradiation on odontoblast-like cells. Laser Phys Lett 5:680–685
Ozawa Y, Shimizu N, Kariya G, Abiko Y (1998) Low-energy laser irradiation stimulates bone nodule formation at early stages of cell culture in rat calvarial cells. Bone 22:347–354
Abramovitch-Gottlib L, Gross T, Naveh D, Geresh S, Rosenwaks S, Bar I, Vago R (2005) Low level laser irradiation stimulates osteogenic phenotype of mesenchymal stem cells seeded on a three-dimensional biomatrix. Lasers Med Sci 20:138–146
Kim HK, Kim JH, Abbas AA, Kim DO, Park SJ, Chung JY, Song EK, Yoon TR (2009) Red light of 647 nm enhances osteogenic differentiation in mesenchymal stem cells. Lasers Med Sci 24:214–222
Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM (2002) Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418:41–49
Horvát-Karajz K, Balogh Z, Kovács V, Drrernat AH, Sréter L, Uher F (2009) In vitro effect of carboplatin, cytarabine, paclitaxel, vincristine, and low-power laser irradiation on murine mesenchymal stem cells. Lasers Surg Med 41:463–469
Peng F, Wu H (2009) Comments on: Red light of 647 nm enhances osteogenic differentiation in mesenchymal stem cells. Lasers Med Sci 24:985–986
Salic A, Mitchison TJ (2008) A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 105:2415–2420
Quarles LD, Yohay LD, Lever LW, Caton R, Wenstrup RJ (1992) Distinct proliferative and differentiated stages of murine MC3T3-E1 cells in culture: an in vitro model of osteoblast development. J Bone Miner Res 7:683–692
Jaiswal N, Haynesworth SE, Caplan AI, Bruder SP (1997) Osteogenic differentiation of purified, culture-expanded human mesenchymal stem cells in vitro. J Cell Biochem 64:295–312
Khadra M, Lyngstadaas SP, Haanaes HR, Mustafa K (2005) Effect of laser therapy on attachment, proliferation and differentiation of human osteoblast-like cells cultured on titanium implant material. Biomaterials 26:3503–3509
Owen TA, Aronow M, Shalhoub V, Barone LM, Wilming L, Tassinari MS, Kennedy MB, Pockwinse S, Lian JB, Stein GS (1990) Progressive development of the rat osteoblast phenotype in vitro: reciprocal relationships in expression of genes associated with osteoblast proliferation and differentiation during formation of the bone extracellular matrix. J Cell Physiol 143:420–430
Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89:755–764
de Souza SC, Munin E, Alves LP, Salgado MA, Pacheco MT (2005) Low power laser radiation at 685 nm stimulates stem-cell proliferation rate in Dugesia tigrina during regeneration. J Photochem Photobiol B 80:203–207
Mvula B, Mathope T, Moore T, Abrahamse H (2008) The effect of low level laser irradiation on adult human adipose derived stem cells. Lasers Med Sci 23:277–282
Posten W, Wrone DA, Dover JS, Arndt KA, Silapunt S, Alam M (2005) Low-level laser therapy for wound healing: mechanism and efficacy. Dermatol Surg 31:334–340
Luo X, Chen J, Song WX, Tang N, Luo J, Deng ZL, Sharff KA, He G, Bi Y, He BC, Bennett E, Huang J, Kang Q, Jiang W, Su Y, Zhu GH, Yin H, He Y, Wang Y, Souris JS, Chen L, Zuo GW, Montag AG, Reid RR, Haydon RC, Luu HH, He TC (2008) Osteogenic BMPs promote tumor growth of human osteosarcomas that harbor differentiation defects. Lab Invest 88:1264–1277
Holleville N, Mateos S, Bontoux M, Bollerot K, Monsoro-Burq AH (2007) Dlx5 drives Runx2 expression and osteogenic differentiation in developing cranial suture mesenchyme. Dev Biol 304:860–874
Komaki M, Karakida T, Abe M, Oida S, Mimori K, Iwasaki K, Noguchi K, Oda S, Ishikawa I (2007) Twist negatively regulates osteoblastic differentiation in human periodontal ligament cells. J Cell Biochem 100:303–314
Komori T (2008) Regulation of bone development and maintenance by Runx2. Front Biosci 13:898–903
Hughes FJ, Collyer J, Stanfield M, Goodman SA (1995) The effects of bone morphogenetic protein-2, -4 and -6 on differentiation of rat osteoblast cells in vitro. Endocrinology 136:2671–2677
Klionsky DJ, Emr SD (1989) Membrane protein sorting: biosynthesis, transport and processing of yeast vacuolar alkaline phosphatase. EMBO J 8:2241–2250
Acknowledgments
The authors thank Chao Chen for his technical assistance. This work was supported by the National Natural Science Foundation of China (contract grant 50477043).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peng, F., Wu, H., Zheng, Y. et al. The effect of noncoherent red light irradiation on proliferation and osteogenic differentiation of bone marrow mesenchymal stem cells. Lasers Med Sci 27, 645–653 (2012). https://doi.org/10.1007/s10103-011-1005-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-011-1005-z