
Abstract
The purpose of this study was to evaluate the possibility of using a semi-automated repetitive DNA sequences-based polymerase chain reaction (rep-PCR) for typing Pseudomonas aeruginosa isolates. rep-PCR profiles obtained by the DiversiLab® system of 84 P. aeruginosa isolates from distinct epidemiological situations were obtained. rep-PCR groupings were in good agreement with the origin of these isolates. Linked rep-PCR profiles were observed for isolates recovered from a same family of cystic fibrosis (CF) patients, for the etiological agents of clustered cases of nosocomial infections, and for some isolates recovered from a same hospital room. rep-PCR and pulsed-field gel electrophoresis SpeI restricted genomic DNA (PFGE-SpeI) profiles were compared. In a few instances, rep-PCR revealed genetic divergences among isolates of a same group of PFGE-SpeI profiles. These divergences could reflect genetic drifts among closely related isolates, as illustrated by those observed between clinical and environmental isolates of a same group of PFGE-SpeI profiles. The interpretation of such differences will require further studies, but the rep-PCR analysis of P. aeruginosa diversity appeared to be an appropriate method to investigate infra-specific genetic relatedness.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pseudomonas aeruginosa is an opportunistic pathogen resistant to many antibiotics and biocides. It is one of the most important etiological agents of nosocomial pulmonary tract infections, especially those occurring among immuno-compromised patients. Sources of P. aeruginosa are variable, but fecal carriage seems to be one of the main causes of their presence in the hospital environment [1]. Hospital facilities like taps and medical devices are often contaminated by this pathogen, and clinical staff or patients are often at the origin of these contaminations [1, 2]. P. aeruginosa can be responsible for acute and chronic pulmonary infections in cystic fibrosis (CF) patients [3].
The comparison of pulsed-field gel electrophoresis SpeI restricted genomic DNA (PFGE-SpeI) is considered to be the main approach to detect closely related isolates of P. aeruginosa, and is, therefore, traditionally used for epidemiological investigations. However, this method presents some disadvantages, since it is time-consuming and technically demanding, requiring specialized gel electrophoresis equipment and qualified personnel [4]. The reproducibility of the analyses between laboratories also needs rigorous and restrictive standardization, and is often criticized [4, 5]. Considering these disadvantages, a challenge for the academics and diagnostic companies is to develop new discriminatory tools that can type bacteria such as P. aeruginosa with the same reliability as PFGE, but faster and more easily. For this purpose, the repetitive sequence-based polymerase chain reaction (rep-PCR) analysis has been proposed as an alternative. This method targets non coding repetitive sequences, which are spread throughout eubacterial genomes and are highly conserved across species [5, 6]. These repetitive sequences can be amplified by PCR, leading to PCR products profiles, visualized by an agarose gel electrophoresis approach, which can be isolate-specific for some species. A technique based on rep-PCR, standardized and partially automated, is now available; the DiversiLab® semi automated rep-PCR system (Bacterial Barcodes, bioMérieux, Athens, GA, USA). However, this method has shown a variable reliability, which appears to be species-related. This approach, thus, requires a validation for each bacterial species prior to its use in a routine clinical laboratory [4]. Here, we present a comparison of automated rep-PCR profiles, obtained by the DiversiLab® system, and profiles obtained by the PFGE approach, for a set of P. aeruginosa isolates representing various clinical and epidemiological situations.
Materials and methods
Bacterial isolates
Eighty-four P. aeruginosa isolates were collected from three hospitals (Table 1): 43 from Albert Michalon Hospital (Grenoble, France), 16 from Croix-Rousse Hospital (Lyon, France), and 25 from Henry Gabrielle Hospital (Lyon, France). Among the 43 isolates from Albert Michalon Hospital, seven (GR32–GR38) were isolated from members of three families (F1–F3) affected by CF and seven came from an epidemiological investigation of a P. aeruginosa-infected patient, of which two were clinical isolates (GR40 and GR44), four came from hospital facilities (GR39, GR41, GR42, and GR45), and one was isolated from an endoscope (GR43). The last 29 isolates from Albert Michalon Hospital were isolated from the sputum of unrelated CF patients. The 16 isolates collected from Croix-Rousse Hospital were clinical isolates recovered from five independent events of clustered cases of nosocomial infections (named O1 to O5). The 25 isolates from Henry Gabrielle Hospital were isolated from tap water or tap nozzle aerators (21 isolates) and from long-stay patients (four isolates) during an epidemiological investigation of the spread and persistence of P. aeruginosa in a hospital unit over a period of three years [1].
PFGE-SpeI typings
All PFGE-SpeI restricted genomic DNA profiles were performed and analyzed as indicated in Lavenir et al. [1]. PFGE-SpeI profiles were interpreted according to the guidelines proposed by Römling et al. and adapted from Tenover et al. [7, 8]. Isolates showing profiles with less than six band changes were defined as ‘linked isolates,’ and for the isolates from Henry Gabrielle, they were grouped into clones or clonal complexes according to Lavenir et al. [1]. Most PFGE-SpeI profiles defined as linked showed less than two DNA band changes. PFGE profiles showing six or more band changes were considered as ‘different,’ and, thus, not to have originated from a recent common ancestor.
Automated rep-PCR DNA fingerprinting
Bacterial isolates were cultured on Columbia agar plates for 24 h at 37°C. Total bacterial DNA was extracted by the UltraClean™ Microbial DNA Isolation Kit (MO Bio Laboratories, Carlsbad, CA, USA) following the manufacturer’s instructions. The yields of extracted total DNA were estimated by NanoDrop quantification (0.25 µM to 0.5 µM). rep-PCR was performed using the DNA Pseudomonas fingerprinting kit (Bacterial Barcodes, bioMérieux, Athens, GA, USA) in a final volume of 25 μL. Thermal cycles included an initial denaturation of 94°C for 2 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 30 s and extension at 70°C for 90 s, and a final extension at 70°C for 3 min. rep-PCR profiles were obtained using the microfluidic DNA chips (Bacterial Barcodes) and an Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA).
rep-PCR fingerprinting profiles were compared by the DiversiLab® (version 3.3) software using the Pearson correlation coefficient (Bacterial Barcodes). The isolates were characterized as ‘linked isolates’ (similarity above 95% and two or less peak changes) or ‘different’ (similarity less than 95% or more than two peak changes).
Results
Randomly picked CF clinical isolates
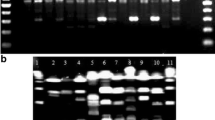
Twenty-nine randomly picked CF clinical isolates from Albert Michalon Hospital were analyzed by the rep-PCR and PFGE-SpeI approaches. Figure 1 shows the rep-PCR profiles obtained by the DiversiLab® system. Significant DNA products ranging from 100 to about 1,000 bp were detected. Most profiles were found to be ‘different’ (more than two band changes), except those of isolates GR17 and GR19 (Fig. 1 and Table 2). PFGE-SpeI analysis showed all isolates to have distinct profiles (data not shown), and were, thus, defined as ‘different’ by this approach (Table 2).

Examples of profiles produced by semi-automated repetitive DNA sequences-based polymerase chain reaction (rep-PCR) among the 29 clinical isolates of Pseudomonas aeruginosa collected in Albert Michalon Hospital. a Profiles of unlinked isolates (index of similitude <95% and more than two peaks different). b Profiles of unlinked isolates (index of similitude >95% but more than two peaks different). c Profiles of linked isolates (index of similitude >95% and exactly two peaks different). Black arrows indicate the peaks that are different between the two profiles
CF isolates from three families
CF P. aeruginosa isolates (n = 7) from three families undergoing medical surveillance at Albert Michalon Hospital were analyzed by the rep-PCR and PFGE SpeI analyses. rep-PCR divided the isolates into two distinct groups. Isolates of families F1 and F2 were defined as related (Table 2). Isolates of the third family F3 (GR36, GR37, GR38) had linked profiles that were found to be distinct from those of families F1 and F2 (Table 2). The PFGE-SpeI analysis confirmed these data. The profiles of isolates from a same family were identical, and those of families F1 and F2 were defined as related.
Epidemiological investigation of a nosocomial outbreak at Albert Michalon Hospital
The source of the etiological agents involved in a nosocomial outbreak at Albert Michalon Hospital was investigated by rep-PCR and PFGE-SpeI analyses. The rep-PCR profiles were found to be different between isolates GR39, GR40, GR42, and GR45. Profiles of isolates GR41, GR43, and GR44 were found to be related, and were defined as linked. The PFGE-SpeI analyses were in line with these results, except that the profile of isolate GR41 was found to be different from those of isolates GR44 and GR43 (Table 2).
Clinical outbreak isolates of the Croix-Rousse Hospital
Isolates (n = 16) from five P. aeruginosa nosocomial outbreaks at Croix-Rousse Hospital were analyzed. The rep-PCR profiles were found to be linked for isolates of each nosocomial outbreak, but to be different between outbreaks (Table 2). PFGE-SpeI typings were in line with these results, but isolates of outbreak O1 (PSE2.17, PSE2.21, PSE2.23, PSE2.25) were found to be similar to those of isolates of outbreak O3 (PSE2.48, PSE2.54, PSE2.63, PSE2.64). The PFGE profiles of these two outbreaks were defined as linked (Table 2).
Isolates from hospital facilities and water networks
Lavenir et al. divided a series of P. aeruginosa isolates from the water network, tap nozzle aerators, and long-stay infected patients into clones and clonal complexes by PFGE-SpeI analysis [1]. Isolates of this study were selected according to these groupings and their origin. Isolates of a same room, of distinct rooms but found to be related by PFGE typings, and from a dominant clone recovered from tap water and tap nozzle aerators of several rooms of the unit, and a case of nosocomial infection at the Henry Gabrielle Hospital, were analyzed. rep-PCR analysis divided the isolates into 11 groups. Isolates from tap water and tap nozzle aerators from room R5 of the hospital unit were divided into three distinct groups, with two isolates (poe92 and poe93) having specific and distinct profiles (Table 2), and all other isolates showing linked profiles. These last few profiles were also observed for isolates recovered from tap water and the tap nozzle aerator of another room (R11). However, the PFGE-SpeI data showed these isolates to be divided into two groups of linked isolates (clonal complex CC2 and CC3). Lavenir et al. demonstrated that the profiles within each group showed a maximum of one DNA band change [1]. The isolates shown to have unique rep-PCR profiles (poe92 and poe93) had PFGE profiles that were grouped with those of isolates poe91 and poe95. All of these isolates had been recovered from tap water and tap nozzle aerators of room R5. The CC3 group clustered profiles from the other isolates of rooms 5 and 11 investigated in this study. For the other isolates, in most cases, a good congruence was observed between the rep-PCR and the PFGE SpeI groupings and the classification into groups of linked or different profiles (Table 2). However, a series of isolates (n = 9) from clonal complex CC22, defined on the basis of PFGE profiles, showed rep-PCR profiles and groupings not in line with the definition of this complex. The rep-PCR analysis divided isolates of CC22 into three entities; two groups of four isolates, and an isolate with a unique profile. Interestingly, the clinical isolate of this complex was found to have a distinct and unique rep-PCR profile. Three other clinical isolates of nosocomial infections were also investigated in this epidemiological investigation. Two of these, poe121 and poe126, were found to be linked by rep-PCR, and the other isolate was found to have a unique profile. These groupings were in line with the PFGE data set, which showed the poe121 and poe126 isolates to have identical profiles.
Discussion
In this study, the use of the semi-automated rep-PCR typing method to investigate various P. aeruginosa epidemiological issues was tested. rep-PCR data sets were compared with those derived from the PFGE-SpeI typing method for a set of P. aeruginosa isolates recovered from randomly picked CF clinical infections, patients of three CF families, infectious outbreaks, and hospital facilities or medical devices. To our knowledge, even if rep-PCR has been already used to type P. aeruginosa strains, this is the first report comparing the efficacy of the semi-automated rep-PCR and the PFGE-SpeI typing methods on isolates of this bacteria [9]. As described in other studies, the semi-automated rep-PCR approach presented many advantages. First of all, the DiversiLab® system was fast [5, 10], allowing an analysis of 13 isolates in a few hours, while the PFGE analysis required two to three days. This rapidity was found to be a major advantage in dealing with epidemiological outbreaks [10]. rep-PCR was also found to be easy to perform and not to require much technical expertise. Most steps in the rep-PCR approach involve limited human intervention (contrary to PFGE). Concerning the analysis of the rep-PCR profiles, the DiversiLab® system provided a user-friendly Internet-based computer-assisted data analysis. Users can, thus, keep track of their data sets, create libraries of profiles, or compare their profiles with those provided by the “Barcodes” library [5, 11]. Combined analysis of new and old data sets can be performed, without additional experiments, whereas for PFGE, DNA profiles often need to be re-run on a same agarose gel. The rules of rejection of relation between isolates during interpretation were easily applied (i.e., similarities less than 95% and more than two peak changes). However, visual inspection of the profiles was found to be essential prior to analysis in order to avoid misinterpretations. A biologist was found to be essential for the final interpretation of the similarities. This important step remains, as for the PFGE SpeI method, a possible source of error.
Concerning the reliability of the semi-automated rep-PCR approach, the choice was made, in this study, to compare the data sets obtained with those derived from PFGE-SpeI analyses. However, these methods make use of totally different strategies. On the one hand, rep-PCR makes use of short-repeated stretches of DNA found among eubacterial genomes, allowing the comparison of rep-containing PCR products ranging in size from about 100 to 1,000 bp. On the other hand, the PFGE-SpeI approach makes use of rare SpeI cutting sites among the P. aeruginosa genomes, allowing the comparison of restricted genomic DNA profiles with DNA bands ranging in size from about 50,000 to 500,000 bp. These two kinds of DNA sequences, rep and SpeI cutting sites, are more than likely to be under distinct selective pressures, with the rep repeats more inclined to be directly involved in recombination events. However, SpeI analysis of restricted genomic DNA will also be affected by recombinational events, but on a larger scale, e.g., a large inversion of a part of a chromosome. It is such genomic instabilities which make these two methods good candidates for an estimation of very closely related isolates which have emerged from a same mother clone just a few days or months ago.
In most instances, a good agreement between the rep-PCR and PFGE-SpeI data sets was observed in this study. However, some discrepancies in the classification of the isolates into linked or different (unique) profiles were observed. These discrepancies between PFGE and rep-PCR data sets that were previously reported by Ross et al. raises the important question of which data set to consider as representative of the true situations [12]. For example, finding clinical, water, and endoscope isolates of a same hospital unit as related by rep-PCR seems quite logical, and can suggest that tap water contamination was at the origin of the endoscope contamination. However, some discrepancies with the PFGE data sets were harder to understand, such as the grouping of isolates from two distinct clonal complexes (e.g., CC2 and CC3). Such a disagreement between the PFGE and rep-PCR analyses could occur because of several reasons. First, it might be the true situation, and reflects the conservation of particular genetic background. Second, the analysis might have been affected by contaminating peaks or ‘bumps,’ not representing true rep-containing PCR products, and creating a misclassification of the isolates. Additional analyses will be required to clarify these discrepancies.
In several epidemiological situations, the rep-PCR data sets appeared to be in line with those derived from PFGE-SpeI analyses. For example, most outbreak isolates were found to be linked to a particular outbreak, and to be different from one outbreak to another by rep-PCR and PFGE. The only exception was a link observed by PFGE between the isolates of two outbreaks. rep-PCR was, thus, shown to be efficient enough to conclude that an outbreak had occurred at the time of observation of these infections. rep-PCR can, thus, be considered as suitable for a real-time follow-up of an outbreak, as proposed for other micro-organisms [4, 5, 13]. It is, thereby, possible to establish the first links between isolates during an outbreak, to exclude unrelated cases, or to rapidly identify contamination sources in order to initiate decontamination as soon as possible.
It also appears that rep-PCR combined with PFGE data sets might bring further insights into the understanding of genetic relatedness among a population of P. aeruginosa. For example, the isolate of a nosocomial infection poe130 was linked by PFGE analysis to a dominant clone of P. aeruginosa found largely distributed among a long-stay hospital unit (CC22). Isolates of this clone were recovered from the tap water and tap nozzle aerators of several rooms of the unit. The rep-PCR data set suggests that these isolates, even though they are part of a same clone (identical PFGE profiles), appear to have undergone some sort of genetic drift, leading to three sub-divisions among this clonal lineage. Most interestingly, the clinical isolate appears to have significantly diverged from the other isolates. This data, thus, suggest that fine genetic modifications might have played a role in the colonization of the patient by this isolate. This will need to be investigated further.
In conclusion, rep-PCR provided results similar to those obtained by the PFGE-SpeI typing method. It was found to be most reliable on P. aeruginosa nosocomial outbreak isolates. For the other situations, its use will need further investigations, but the inferences based on the rep-PCR data sets did not appear to be aberrant. rep-PCR could also bring further insights on the genetic proximity of isolates belonging to a same clone or clonal complex defined by the PFGE approach. However, the reasons for the genetic divergences observed in these last few cases will, again, require further validation by DNA sequence analysis of the rep-PCR DNA products. Such studies will bring a novel view on the rep-PCR data sets that could improve the interpretation guidelines of the profiles.
References
Lavenir R, Sanroma M, Gibert S et al (2008) Spatio-temporal analysis of infra-specific genetic variations among a Pseudomonas aeruginosa water network hospital population: invasion and selection of clonal complexes. J Appl Microbiol 105:1491–1501. doi:10.1111/j.1365-2672.2008.03907.x
Petignat C, Francioli P, Nahimana I et al (2006) Exogenous sources of Pseudomonas aeruginosa in intensive care unit patients: implementation of infection control measures and follow-up with molecular typing. Infect Control Hosp Epidemiol 27:953–957. doi:10.1086/506409
Nazaret S, Assade F, Brothier E et al (2009) RISA-HPLC analysis of lung bacterial colonizers of cystic fibrosis children. J Microbiol Methods 76:58–69. doi:10.1016/j.mimet.2008.09.019
Harrington SM, Stock F, Kominski AL et al (2007) Genotypic analysis of invasive Streptococcus pneumoniae from Mali, Africa, by semiautomated repetitive-element PCR and pulsed-field gel electrophoresis. J Clin Microbiol 45:707–714. doi:10.1128/JCM.01871-06
Healy M, Huong J, Bittner T et al (2005) Microbial DNA typing by automated repetitive-sequence-based PCR. J Clin Microbiol 43:199–207. doi:10.1128/JCM.43.1.199-207.2005
Maslow JN, Mulligan ME, Arbeit RD (1993) Molecular epidemiology: application of contemporary techniques to the typing of microorganisms. Clin Infect Dis 17:153–162
Tenover FC, Arbeit RD, Goering RV et al (1995) Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol 33:2233–2239
Römling U, Wingender J, Müller H et al (1994) A major Pseudomonas aeruginosa clone common to patients and aquatic habitats. Appl Environ Microbiol 60:1734–1738
Tam VH, Chang KT, LaRocco MT et al (2007) Prevalence, mechanisms, and risk factors of carbapenem resistance in bloodstream isolates of Pseudomonas aeruginosa. Diagn Microbiol Infect Dis 58:309–314. doi:10.1016/j.diagmicrobio.2007.05.006
Carretto E, Barbarini D, Farina C et al (2008) Use of the DiversiLab® semiautomated repetitive-sequence-based polymerase chain reaction for epidemiologic analysis on Acinetobacter baumannii isolates in different Italian hospitals. Diagn Microbiol Infect Dis 60:1–7
Wise MG, Healy M, Reece K et al (2007) Species identification and strain differentiation of clinical Candida isolates using the DiversiLab® system of automated repetitive sequence-based PCR. J Med Microbiol 56:778–787. doi:10.1099/jmm.0.47106-0
Ross TL, Merz WG, Farkosh M et al (2005) Comparison of an automated repetitive sequence-based PCR microbial typing system to pulsed-field gel electrophoresis for analysis of outbreaks of methicillin-resistant Staphylococcus aureus. J Clin Microbiol 43:5642–5647. doi:10.1128/JCM.43.11.5642-5647.2005
Syrmis MW, O’Carroll MR, Sloots TP et al (2004) Rapid genotyping of Pseudomonas aeruginosa isolates harboured by adult and paediatric patients with cystic fibrosis using repetitive-element-based PCR assays. J Med Microbiol 53:1089–1096. doi:10.1099/jmm.0.45611-0
Acknowledgments
The authors acknowledge Dr. S. Tigaud at Croix-Rousse Hospital for the initiation of this study and L. Loiseau for her technical assistance.
The authors would like to thank the Cluster “Environnement” of the Rhône-Alpes Region and the CNRS for their financial support of the PAR-MIC technical platform (UMR5557 Microbial Ecology, Villeurbanne, France). This work was partly supported by the Agence Nationale de la Recherche (ANR) SEST 2005 009-01 and CESA 2008 022-01 projects.
Author information
Authors and Affiliations
Corresponding author
Additional information
A. Doléans-Jordheim and B. Cournoyer contributed equally to this work.
Rights and permissions
About this article
Cite this article
Doléans-Jordheim, A., Cournoyer, B., Bergeron, E. et al. Reliability of Pseudomonas aeruginosa semi-automated rep-PCR genotyping in various epidemiological situations. Eur J Clin Microbiol Infect Dis 28, 1105–1111 (2009). https://doi.org/10.1007/s10096-009-0755-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-009-0755-z