
Abstract
As invasive fungal infections in immunocompromised patients become increasingly important, the field of antifungal chemotherapy continues to evolve rapidly. New agents have entered the clinical arena, providing physicians with a variety of choices for treatment of most infections. Standardized methods for testing the in vitro susceptibility of fungi have become available, and concentration-effect relationships are increasingly explored. Finally, the availability of an entirely new class of antifungal agents is opening new opportunities for combination therapy of infections that are notoriously difficult to treat and carry a dismal prognosis. However, the ongoing progress in these key areas has also made antifungal chemotherapy considerably more complex and susceptible to misconceptions. Continuing efforts in the laboratory and well designed collaborative clinical trials are needed more than ever to turn opportunities into lasting benefit for patients at risk for or suffering from life-threatening invasive mycoses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past two decades, fungi have emerged as important causes of infectious morbidity and mortality in immunocompromised patients. The most significant risk factors include profound and prolonged granulocytopenia, immunosuppression with corticosteroids, acquired deficiencies in the number and/or function of T-helper cells, and severe illness requiring multiple invasive medical procedures, such as the use of intravascular devices and extensive abdominal surgery. While Aspergillus fumigatus and Candida albicans traditionally account for the majority of invasive opportunistic infections, more recent epidemiological trends indicate a shift toward infections by non-fumigatus Aspergillus spp., non-albicans Candida spp., and previously uncommon fungi that often display resistance to current antifungal agents in vitro and in vivo [1]. Human immunodeficiency virus (HIV)-infected patients with advanced immune dysfunction are particularly susceptible to cryptococcal meningitis, disseminated histoplasmosis, coccidioidomycosis, and penicillosis, and recent outbreaks highlight that endemic fungi can become a significant public health concern beyond their baseline prevalence [2, 3].
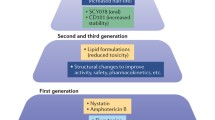
For many years, the treatment of invasive fungal infections was limited to amphotericin B deoxycholate with or without the addition of 5-fluorocytosine. It was not until the late 1980s that the first durably useful alternatives emerged through the advent of fluconazole and itraconazole. Prompted by the exponential increase of severely immunocompromised patients at risk for invasive fungal infections, however, the past 10 years have witnessed a major expansion in antifungal drug research, as reflected by the introduction of less toxic lipid formulations of amphotericin B as well as the ongoing development of novel echinocandin lipopeptides and improved antifungal triazoles [4] (Fig. 1). Improved blood culture, antigen, and nucleic acid detection techniques [5, 6, 7], the advent of high-resolution two-dimensional imaging [8, 9], and an increased awareness among physicians of the fungal pandemic have all had considerable impact on improving the early clinical diagnosis of invasive fungal infections, and major advances have been achieved in harmonizing disease definitions, in defining paradigm for antifungal interventions, and in designing and implementing clinical trials [10, 11, 12]. Last, but not least, standardized methods for testing the in vitro susceptibility of fungi have become available and are continuously refined [13], and concentration-effect relationships in vitro and in vivo are increasingly explored [14, 15]. Nevertheless, despite these advances, invasive fungal infections remain difficult to diagnose and to manage, and there is a continuing need for improved antifungal therapy.

Schematic diagram of current antifungal agents and their targets in the fungal cell. *, investigational agents
Current Role of Antifungal Susceptibility Testing
Establishing reproducible in vitro methods to assess antimicrobial susceptibility is an important tool for the identification of microbiologically resistant organisms and for optimal selection of antimicrobial therapy. The experience with antibacterial chemotherapy indicates superior outcomes for therapies that are guided by the results of in vitro susceptibility testing as opposed to a merely species-based therapy [16]. Standardized methods for testing the in vitro susceptibility of yeasts [17, 18] and filamentous fungi [19] to current antifungal agents have become available. Tentative breakpoints have been established for fluconazole, itraconazole, and 5-fluorocytosine against Candida spp. [13, 17]; for azole antifungal agents against Candida spp., these breakpoints appear to have predictive utility similar to that observed with in vitro susceptibility testing of antibacterial agents [20]. However, for other organism-drug combinations, correlation of in vitro susceptibility with antifungal activity in vivo remains difficult to establish [20, 21]. This is partly related to ongoing methodological problems associated with the selection of optimal assay conditions and endpoints, particularly for the polyenes and the echinocandins, but also is due to the prominent role of host- and disease-related factors in the outcome of most invasive opportunistic fungal infections.
Unlike pathogenic bacteria, in which resistance may emerge rapidly and spread, fungi do not become rapidly resistant because of their eukaryotic nature, their longer replication time, and their lack of genetic mechanisms for the exchange of resistance and of drug-degrading substances. Currently, the emergence of resistance is essentially limited to the following two scenarios [22]: (i) primary emergence of a naturally resistant species, such as Trichosporon asahii or Pseudallescheria boydii, that is resistant to the fungicidal activity of amphotericin B; and (ii) the selection of a resistant species during antifungal therapy, as exemplified by breakthrough infections with Candida krusei or Candida glabrata during systemic prophylaxis with triazoles [4]. Stepwise, cumulative molecular events that lead to progressively decreased susceptibility and stable resistance during exposure to current azoles are rarely encountered in patients but have been reported following longstanding exposure to azoles in conjunction with HIV-associated oropharyngeal candidiasis [23, 24] and, less well characterized, chronic granulomatous disease [25].
As the use of antifungal azoles in medicine, agriculture [26], and animal health [27] becomes more widespread, the selection and nosocomial spread of azole-resistant Candida spp. appears inevitable (Fig. 2). To meet this challenge, a thorough understanding of the molecular mechanisms of antifungal drug resistance is required. During the past few years, increased target expression, alterations at the target binding site, and the presence of inducible efflux pumps have been identified as mechanisms of azole resistance and may offer targets for intervention [28, 29]. Comparatively little is known about resistance mechanisms against polyenes and echinocandins; changes in the composition of the fungal cell wall and in the sterol chemistry of the cell membrane have been described for fungi exposed to amphotericin B [24, 30], and mutations of the FKS1 gene have been observed in fungi exposed to echinocandins [31].

Evolution of antimicrobial resistance. Selective pressure arises upon prolonged exposure to a given habitat in which the microorganism in question is prevalent. Induction of resistance is dependent on the genetic versatility of the microorganism, and evolutionary success of resistant clones relies on their biological fitness and opportunities for nosocomial transmission
In clinical practice, the microbiological diagnosis should be attempted as feasible in all cases of suspected invasive fungal infection, with the organism identified to the species level. Because of the lack of its predictive value in other settings, in vitro susceptibility testing is currently limited to Candida spp. versus fluconazole and flucytosine, respectively. However, additional in vitro testing of other organism/drug combinations may be indicated in refractory infections and within surveillance programs [20, 21] (Fig. 3).

Proposed approach to identification and antifungal susceptibility testing of fungal organisms for selection of antifungal therapy (modified from Rex and Pfaller [20])
Pharmacodynamics of Antifungal Compounds
In a broad sense, the term pharmacodynamics encompasses the description of concentration-over-time relationships of antifungal drugs and drug combinations in vitro and in vivo. Common, nonstandardized tools to study the pharmacodynamics of antifungal drugs in vitro are listed in Fig. 4. While these methods provide important information on the mode of action of antifungal drugs, they have major technical and biological limitations. Changing assay conditions such as inoculum size, medium, or pH may result in conflicting observations and, therefore, uncertainty as to their therapeutic relevance. Biological factors, such as different growth characteristics of the organism in vivo and the absence of host defense factors, plasma pharmacokinetics and tissue distribution, and protein binding and carrier effects, are further impediments to an immediate translation to the therapeutic setting. Therefore, results and observations from pharmacodynamic in vitro studies should always be interpreted with caution and further investigated in appropriately designed animal models.

Overview of currently available methods to assess concentration-time-effect relationships of antifungal agents in vitro
One of the principal aims of antimicrobial drug therapy is the characterization of the relationships between dose, dosage interval, drug concentrations in the body, in vitro susceptibility of the microorganism, and drug effects. Understanding these pharmacokinetic-pharmacodynamic relationships provides important knowledge of a drug’s mode of action and can be instrumental in setting susceptibility breakpoints and in guiding optimal dosing regimens [32]. Due in large part to the importance of host- and disease-related factors for patient outcome and the lack of reliable surrogate markers in invasive mycoses, the evaluation of pharmacokinetic and pharmacodynamic relationships relies on well-controlled infection models that provide true endpoints. Such models, by virtue of pharmacodynamic and dose-fractionating studies, allow for the exploration of the relationships of pharmacodynamic parameters such as Cmax/MIC, AUC/MIC, and the length of time that plasma concentrations stay above the MIC (Ttau>MIC) with antifungal efficacy (Fig. 5). Contemporary approaches to treating most invasive mycoses are based on doses and dosage schedules that have been empirically derived over time. Experimental exploration of pharmacokinetic-pharmacodynamic relationships has begun only recently [14, 15], and incorporation of pharmacodynamic endpoints into clinical studies remains an important goal.

Commonly used pharmacodynamic parameters to assess pharmacokinetic-pharmacodynamic relationships of antifungal agents in vivo. Correlation of these parameters with endpoints of outcome through mathematical equations in experimental models of invasive fungal infections allows for determination of 50% or 90% effective parameter values. In addition, fractionating the identical daily dose in 1–4 divided doses and of comparison of pharmacodynamic parameters and therapeutic effect among the different cohorts may enable identification of the parameter most predictive of therapeutic success. Pharmacodynamic modelling and dose-fractionating studies can be used as guidance for setting susceptibility breakpoints and for selection of the optimal dose and dosage schedule in patients. Cmax/MIC, ratio of peak plasma level and MIC; AUC/MIC, ratio of the area under the concentration-vs.-time curve and the MIC; Ttau>MIC, time during the dosing interval that plasma concentrations exceed the MIC for the investigated isolate(s)
Although the target sites of current antifungal drug classes are quite limited, the existence of these targets and the pharmacodynamic consequences of the drug-target interaction are quite diverse due to the enormous variety of fungal organisms that are biologically very different. In the following paragraphs, we will briefly review mechanism of action, antifungal spectrum, concentration-effect relationships in vitro, and pharmacokinetic/pharmacodynamic relationships of the existing classes of antifungal compounds, along with resistance of fungi to these compounds, with a focus on clinical implications and open questions.
I. Antifungal Polyenes
Class and Mechanism of Action
The antifungal polyenes consist of a family of some 200 natural macrolide antibiotics, of which only amphotericin B and nystatin have been developed for systemic therapy. There are currently four licensed amphotericin B formulations: amphotericin B deoxycholate, amphotericin B colloidal dispersion, amphotericin B lipid complex, and a small unilamellar liposomal amphotericin B. The lipid carriers of these formulations have distinct physicochemical and pharmacokinetic characteristics. However, it is largely unknown whether the marked differences in pharmacokinetics also have a clinically relevant impact on the pharmacodynamics of these agents [33, 34, 35]. A multilamellar liposomal formulation of nystatin entered clinical trials in the 1990s [4, 36] but has not been further developed.
The principal mechanism of action of the polyenes is specific binding to ergosterol in the fungal cell membrane. This binding results in the disorganization of the membrane, possibly by formation of specific pores composed of small aggregates of drug and ergosterol. These defects cause depolarization of the membrane, an increase in membrane permeability to protons and monovalent cations, and eventually, cell death. The polyenes also bind to other sterols such as cholesterol, although with less avidity; nevertheless, this accounts for much of their toxicity. A contributory mechanism of action of amphotericin B may be the generation of oxidative metabolites, possibly due to auto-oxidation of the compound with formation of free radicals or an increase in membrane permeability [4, 37].
Spectrum of Antifungal Activity, and Resistance of Fungal Pathogens
Amphotericin B has broad-spectrum antifungal activity that includes most opportunistic and endemic fungi. Notable exceptions are Candida lusitaniae, Aspergillus terreus, and some of the emerging pathogens such as Trichosporon asahii, Fusarium spp., Pseudallescheria boydii, Scedosporium prolificans, and Paecilomyces lilacinus [1, 4]. Given the still-evolving methodology for resistance testing of amphotericin B in vitro [20], it is unclear whether primary resistance among fungal pathogens is truly uncommon or just difficult to detect. Primary microbiological resistance to amphotericin B appears to be due predominantly to quantitative or qualitative alterations in membrane-associated ergosterol. The description of secondary resistance is restricted to anecdotal cases of patients who received nonresorbable polyenes as antifungal prophylaxis [21, 30].
Pharmacokinetics
After intravenous administration of the conventional formulation in deoxycholate, the compound dissociates from its lipid carrier, becomes highly (>95%) protein bound, and distributes preferentially into organs of the mononuclear phagocytic system. The drug follows a biphasic pattern of elimination from plasma with an initial (beta-) elimination half-life of 24–48 hours, followed by a long terminal (gamma-) half-life of several days. The drug is slowly eliminated into urine and bile, with 62% of a dose recovered in unchanged form in urine and feces at 1 week. No metabolites have been identified thus far, and recent mass balance studies suggest that metabolism plays at most a minor role in amphotericin B elimination [4, 38, 39].
In comparison to amphotericin B deoxycholate, the so-called amphotericin B lipid formulations (amphotericin B colloidal dispersion, amphotericin B lipid complex, and the small unilamellar liposomal amphotericin B) have reduced nephrotoxicity yet they preserve the antifungal activity of the parent. Whereas amphotericin B colloidal dispersion (Amphotec) is not fundamentally different from conventional amphotericin B with regard to plasma pharmacokinetics, amphotericin B lipid complex (Abelcet) is more rapidly taken up by the mononuclear phagocytic system, and the small unilamellar liposomal formulation (AmBisome) achieves comparatively higher peak plasma levels and a prolonged and stable circulation in plasma [33, 34, 39, 40]. Independent of its formulation and on the basis of its prolonged half-life in plasma, amphotericin B is usually administered once daily. Due to their reduced nephrotoxicity, the lipid formulations allow for the delivery of higher doses than with conventional amphotericin B. However, the majority of animal models have also demonstrated that higher doses are usually required to achieve antifungal efficacy equivalent to that of conventional amphotericin B [34].
In Vitro Pharmacodynamics
Investigations on the impact of drug concentrations on the rate and extent of organism killing in vitro by time-kill studies consistently revealed concentration-dependent fungicidal activity of amphotericin B against Candida albicans and Cryptococcus neoformans [41, 42]. As drug concentrations are increased, both the rate and extent of antifungal activity is enhanced. However, the in vitro fungicidal properties of amphotericin B are organism-dependent. While amphotericin is highly fungicidal against Candida albicans, the drug is not fungicidal against various emerging fungal pathogens at safely achievable concentrations. An example is Trichosporon asahii, an uncommon but life-threatening cause of disseminated infection in granulocytopenic patients [43]. In vitro pharmacodynamic studies have demonstrated that amphotericin B inhibits but does not kill Trichosporon, and these findings correlated with lack of fungicidal activity in experimental disseminated trichosporonosis and clinical resistance to maximum tolerated doses [44, 45]. Consistent with its mechanism of action, amphotericin B also exerts prolonged postantifungal effects (PAFEs) in vitro, ranging from 0.5 to 10.6 hours and from 2.8 to 10.4 hours for Candida albicans and Cryptococcus neoformans, respectively [46]. More recent data with the same fungal species demonstrate PAFEs of greater than 12 hours with amphotericin B concentrations above the MIC and shorter PAFEs of 0–2 hours for concentrations below the MIC [47].
In Vivo Pharmacodynamics
Experimental pharmacodynamic in vivo studies support the concentration-dependent killing effects observed in vitro. In a dose-fractionating study in neutropenic mice with disseminated candidiasis, animals received total doses ranging from 0.8 to 20 mg/kg over 72 hours divided into 1, 3, or 6 fractions. The peak/MIC ratio was the parameter that provided the best relationship with the residual fungal burden in kidney tissue (r 2=0.93), followed by time above the MIC (Ttau>MIC; r 2=0.82) and the AUC/MIC ratio (r 2=0.61). This study also demonstrated prolonged in vivo PAFEs ranging from 23 to 30 hours [48]. In a Candida albicans neutropenic murine lung infection model investigating the effects of escalating doses of liposomal amphotericin B in single or divided daily doses, single daily high doses (20–30 mg/kg/day) had a greater effect on fungal burden than lower- or divided-dose regimens [49]. Similarly, in a rabbit model of central nervous system candidiasis examining all four licensed amphotericin B formulations, a strong inverse correlation was observed between fungal burden in brain tissue and plasma concentrations of total amphotericin B, with higher concentrations demonstrating a more pronounced effect [50].
The distinct pharmacokinetic and pharmacodynamic properties of the lipid formulations have been used to investigate dose escalation of amphotericin B in patients with invasive fungal infections. In a formal maximum tolerated dose study, escalating doses of liposomal amphotericin B were investigated in 21 patients with proven or probable aspergillosis, zygomycosis, or fusariosis [51]. Doses as high as 15 mg/kg were well tolerated, and 68% of patients achieved a successful outcome by intent-to-treat analysis. Similarly, the accumulation of large concentrations of amphotericin B in the mononuclear phagocytic system achieved by amphotericin B lipid complex [52] was harnessed to explore a strategy for treatment of hepatosplenic candidiasis. Loading of tissues with amphotericin B lipid complex in the amount of 100 mg/kg over 6 weeks resulted in a continued resolution of hepatic and splenic lesions for 6 months after discontinuation of therapy [53].
Clinical Implications
The collective evidence from these studies implies an important consideration for the use of amphotericin B in clinical practice. The concentration-dependent fungicidal effects, the prolonged PAFEs, and the dose- and concentration-dependent antifungal efficacy in experimental models of invasive fungal infections (Tables 1 and 2) all suggest that large, daily doses will be most effective and that achievement of optimal peak concentrations is important. As a consequence, the dose of amphotericin B should not be reduced injudiciously, and infusion for longer durations than approved by the regulatory authorities should be avoided. Finally, dose escalation appears to be a valid strategy for treatment of clinically refractory infections by amphotericin B-susceptible organisms that should be further pursued.
II. Flucytosine
Mechanism of Action
Flucytosine (5-fluorocytosine) is a low-molecular-weight, synthetic, fluorinated, pyrimidine analogue. Following uptake by the fungus-specific enzyme cytosine permease, it is converted to 5-fluorouracil, a potent anticancer agent that causes RNA miscoding and inhibits DNA synthesis [54].
Spectrum of Antifungal Activity, and Resistance of Fungal Pathogens
Flucytosine has broad-spectrum antifungal activity against Candida spp., Cryptococcus neoformans, Saccharomyces cerevisiae, and certain dematiaceous moulds [55, 56]. Resistance to 5-fluorocytosine in susceptible species may involve either mutations in enzymes necessary for cellular uptake, transport, or metabolism, or competitive upregulation of pyrimidine synthesis [57]. Primary resistance in invasive isolates of Candida albicans and Cryptococcus neoformans is currently reported in 0–8% [29] and <2% [58], respectively, and is due mostly to defects in cytosine deaminase. Secondary resistance is caused primarily by defects in the downstream intracellular metabolism and has been observed in up to 40% of patients receiving flucytosine monotherapy [21]. Because of the potential for development of secondary resistance, flucytosine is rarely given alone but in combination with amphotericin B or, more recently, fluconazole. Since marked increases in the frequency of resistance in vitro have been observed below 25 μg/ml, it has been recommended that serum levels in patients be maintained above that concentration [57].
Pharmacokinetics
Flucytosine is readily absorbed from the gastrointestinal tract, has negligible protein binding, and distributes evenly into tissues and body fluids. It undergoes only minor hepatic metabolism and is eliminated predominantly in active form into the urine. On the basis of an elimination half-life of 3–6 hours in patients with normal renal function, flucytosine is usually administered in 3–4 equally divided doses [4]. Close monitoring of plasma levels and dosage adjustment is recommended, particularly with impaired renal function. Peak plasma levels between 40 and 60 mg/l correlate with antifungal efficacy and are rarely associated with hematopoietic toxicity [59]. The drug is available in oral and parenteral form, but only the oral formulation is approved in the USA.
In Vitro Pharmacodynamics
Time-kill assays performed against Candida spp. and Cryptococcus neoformans suggest a predominantly concentration-independent fungistatic (≤99% reduction in cfu) activity of flucytosine at clinically achievable plasma concentrations [60, 61], i.e. the rate and extent of activity is generally not increased when concentrations of flucytosine exceed the MIC for the isolates. Prolonged PAFEs against Candida spp. and Cryptococcus neoformans have been consistently noted. Growth suppression was dependent on concentration and duration of exposure and ranged from 0.8 hours to up to 10 hours [61, 62].
In Vivo Pharmacodynamics
The pharmacokinetic and pharmacodynamic relationships of flucytosine have been investigated using dose fractionating in a neutropenic kidney target mouse model of disseminated candidiasis. Increasing doses produced minimal concentration-dependent killing; nevertheless, flucytosine produced a dose-dependent suppression of growth after plasma concentrations had fallen below the MIC. The fungistatic dose increased with lengthening dosing interval. Nonlinear regression analysis revealed that both the time above the MIC and the AUC/MIC ratio were important in predicting efficacy, whereas the peak level/MIC ratio was the least important parameter. Maximum efficacy was observed when levels exceeded the MIC for only 20–25% of the 24-hour dosing interval [63].
Clinical Implications
The published in vitro and in vivo data suggest that flucytosine exhibits concentration-independent or time-dependent activity (Tables 1 and 2). Since current dosing regimens achieve plasma levels that stay severalfold above the MIC90 of Candida albicans isolates for periods exceeding the usual dosing interval of 6 hours, the use of lower doses or less frequent dosing may be worthwhile to explore [15]. New dosing regimens may yield identical antifungal efficacy while further reducing potential toxicities that are believed to be concentration dependent [59].
III. Antifungal Triazoles
Mechanism of Action
The antifungal triazoles (fluconazole, itraconazole, voriconazole, and investigational agents posaconazole and ravuconazole) are synthetic compounds that have one or more azole rings with three nitrogen atoms in the five-member ring. They act predominantly by inhibition of the CYP450-dependent conversion of lanosterol to ergosterol. This leads to an accumulation of toxic 14-a-methylsterols and a depletion of membrane-associated ergosterol, which alters cell membrane properties and function and results in inhibition of cell growth and replication [4].
Spectrum of Antifungal Activity, and Resistance of Fungal Pathogens
The triazoles are active against dermatophytes, Candida albicans, non-albicans Candida spp., Cryptococcus neoformans, and the dimorphic (endemic) fungi [64, 65, 66]. Among non-albicans Candida spp., they are less active against Candida glabrata and, with the exception of voriconazole and the investigational triazoles, inactive against Candida krusei [20]. Clinically useful activity against Aspergillus spp. and dematiaceous moulds is restricted to itraconazole and voriconazole [67]. Voriconazole also has activity against Fusarium spp. [66]. The mechanisms of resistance to antifungal azoles include differences or alterations in the composition of membrane-associated sterols, alterations in the biosynthetic pathway of ergosterol, genetic changes in the target enzyme (mutation, overexpression, gene amplification), and enhanced efflux by ABC transporters and major facilitators [24, 29]. Secondary resistance has been observed following prolonged exposure to azoles in the settings of chronic recurrent oropharyngeal candidiasis, allogeneic hematopoietic stem-cell transplantation, and chronic granulomatous disease and is thought to be primarily due to the selection of resistant clones. Stepwise induction of stable secondary resistance, however, has been observed in HIV-infected patients with chronic recurrent oropharyngeal candidiasis [23, 24], highlighting the potential epidemiological threats by injudicious azole prophylaxis.
Pharmacokinetics
The antifungal triazoles are generally well tolerated, but they have significant potential for drug-drug interactions through their interference with cytochrome P450-dependent oxidative metabolism.
The three currently approved compounds have quite different pharmacokinetic properties. While fluconazole has nearly complete oral bioavailability, circulates in plasma mostly as free drug, undergoes only negligible hepatic metabolism, and is excreted predominantly through the kidneys in unchanged form, itraconazole, in contrast, is not as well absorbed by the gastrointestinal tract, displays high protein binding extensive hepatic metabolism, and is excreted in inactive form via the liver and kidneys [4]. Voriconazole has good oral bioavailability but exhibits nonlinear pharmacokinetics. Protein binding is low, and tissue and CSF levels exceed those of trough plasma levels severalfold. Elimination occurs primarily by oxidative hepatic metabolism, and only small amounts of voriconazole are excreted in unchanged form into the urine. Unlike fluconazole and itraconazole, where CYP3A4 is the predominant isoenzyme, oxidative metabolism of voriconazole occurs primarily by CYP2C19, although CYP2C9 and CYP3A4 also contribute. Of note, there is wide between-subject variability in disposition that is related to genetic CYP2C19 polymorphism [66, 68].
It is unclear whether the differences in the disposition of the current triazoles may result in different efficacy at different sites. Also unclear is the clinical impact of genetic polymorphisms in the hepatic metabolization of voriconazole. All agents are available as oral and parenteral formulations. On the basis of their extended half-lives, fluconazole and intravenous itraconazole are approved for once-daily dosing; the oral formulations of itraconazole as well as voriconazole are administered on a twice-daily schedule.
Fluconazole
In Vitro Pharmacodynamics
On the basis of in vitro observations, fluconazole is generally considered to be a fungistatic agent [14, 15]. Time-kill studies for susceptible Candida albicans, Candida tropicalis, and Candida glabrata conducted over an incubation period of 24 hours at multiples of the MIC showed fungistatic activity (<99.9% decrease in the log cfu/ml compared with starting inoculum), with minimal concentration-related growth effects observed between concentrations equal to 0.5 times and 2 times the MIC, but not beyond [41]. Against three susceptible clinical isolates of Cryptococcus neoformans, similar pharmacodynamics were observed at concentrations ≥0.5 times the MIC, with the rate and extent of antifungal activity being concentration independent [42]. However, time-kill studies with Candida albicans in which extended incubation periods and nonproliferating growth conditions were used demonstrated direct fungicidal activity of fluconazole against non-proliferating Candida albicans [69]. While the underlying mechanism is unclear, these findings raise the possibility that fluconazole might eliminate Candida spp. over extended periods without help from host defenses. This would be consistent with the efficacy of fluconazole in the treatment of disseminated candidiasis in persistently neutropenic rabbits [70] and in neutropenic patients [71, 72].
At various concentrations and exposure times and in different serum-free growth media, fluconazole displayed no measurable PAFE against Candida albicans or Cryptococcus neoformans [47, 73]. However, when the assays were performed in the presence of fresh serum, concentration-dependent PAFEs against Candida albicans in the range of 1.1–3.6 hours were noted; pretreatment also increased the vulnerability of Candida albicans to killing by polymorphonuclear leukocytes [73].
In Vivo Pharmacodynamics
Consistent with the results of time-kill assays, fluconazole demonstrated fungistatic (<99.9% reduction in cfu/ml) activity against Candida albicans in a dynamic 48 hour one-compartment in vitro bloodstream infection model; doubling of the fluconazole dose did not increase rate or extent of activity against either of the two isolates [74]. The pharmacodynamics of fluconazole in vivo have been investigated in murine models of disseminated candidiasis that used the fungal burden in kidney tissue as endpoint for antifungal efficacy. In a non-neutropenic mouse model, dose fractionating revealed similar reductions in fungal densities between groups that received the same total dose of fluconazole in one, two, or four equally divided doses. Calculation of pharmacodynamic parameters suggested that the pharmacodynamic parameter of fluconazole that best predicted outcome was the AUC/MIC ratio [75]. These findings were corroborated in neutropenic mice using three Candida albicans strains for which MICs of fluconazole spanned the range of susceptible and susceptible dose-dependent MICs (0.5, 16, and 32 µg/ml). The magnitude of the AUC/MIC ratio required to reach 50% of the effective dose (ED50) was similar for all three organisms and ranged from 12 to 25 [76].
Clinical Implications
Investigations of pharmacokinetic and pharmacodynamic relationships in patients have not been presented to date. The dose-independent pharmacokinetics (Tables 1 and 2) as well as the available experimental and clinical data are in support of once-daily dosing regimens. Current susceptibility breakpoints and dosing recommendations for fluconazole against Candida spp. have been derived from MIC and outcome information of a limited set of patients with mostly superficial Candida infections [13]. The principal feasibility of this approach is supported by several animal studies that have demonstrated a correlation between MIC and antifungal efficacy [14] and supporting evidence from clinical studies [20]. However, given the still-evolving state of antifungal susceptibility testing methodologies and the limited clinical database with which it is correlated, treatment of serious invasive mycoses caused by organisms for which MICs are in the susceptible dose-dependent range by dose escalation warrants controlled investigation before its injudicious incorporation into clinical practice.
Itraconazole
In Vitro Pharmacodynamics
Itraconazole exerts species- and strain-dependent fungicidal activity against filamentous fungi [77, 78, 79] and, with the exception of some strains of Cryptococcus neoformans [80], is generally fungistatic in vitro against yeast-like fungi. The biological background for this differential mode of action is not clear.
Time-kill experiments in conventional media have demonstrated concentration-independent, fungistatic activity of itraconazole against Candida spp. and Cryptococcus neoformans, with maximum effectiveness observed at two times the MIC and four to eight times the MIC, respectively [72, 81]; performance of time-kill assays in the presence of serum (80%) did not alter the dynamics observed [82]. Against Aspergillus spp., itraconazole displayed time- and concentration-dependent fungicidal activity with >87–>97% killing within 24 hours of drug exposure [72]. Studies on persistent effects or using in vitro pharmacodynamic models have not been published to date.
In Vivo Pharmacodynamics
The principal possibility of a correlation between in vitro susceptibility data and outcome has been demonstrated for itraconazole in a murine model of disseminated aspergillosis [83]. In a model of invasive pulmonary aspergillosis in methylprednisolone/cyclosporine-immunosuppressed rabbits receiving oral itraconazole, there was a significant pharmacodynamic relationship (r=0.87, P<0.001) between itraconazole concentrations in plasma and antifungal efficacy as a function of the burden of Aspergillus fumigatus in lung tissue [84]. In persistently neutropenic rabbits with invasive pulmonary aspergillosis receiving intravenous itraconazole, best correlations were found between AUC, Cmax, and Cmin, suggesting concentration- and time-dependent pharmacodynamic relationships [85]. These studies provide the experimental support for the concept of a critical threshold concentration as a surrogate marker for prediction of the drug’s concentration and efficacy in the respiratory tract.
Clinical Implications
The main rationale for monitoring plasma levels in patients has been the highly variable bioavailability of oral itraconazole. Initially, the target plasma level for oral itraconazole was set at 0.25 µg/ml (high-performance liquid chromatography) at trough on the basis of MIC90s in vitro; later, data on concentrations in patients who received oral itraconazole for prophylaxis suggested the maintenance of plasma levels between 0.25 and 0.5 µg/ml [86, 87]. More recently, the predictive value of threshold concentrations has been analyzed in a cohort of 190 patients with acute leukemia who received antifungal prophylaxis with oral itraconazole. A total of 27 patients developed a proven or probable invasive mould (n=25) or yeast (n=2) infection. The median itraconazole trough concentration after the first week of prophylaxis was significantly lower in patients who developed invasive fungal infections (0.46 vs. 0.82 µg/ml, P=0.008). Multivariate logistic regression analysis demonstrated a significant (P=0.028) statistical association of trough concentrations <0.5 µg/ml with the occurrence of invasive fungal infections [88]. This study strongly suggests that minimum trough levels of ≥0.5 µg/ml (high-performance liquid chromatography) should be achieved and maintained when oral itraconazole is given to neutropenic patients for prophylaxis.
In the therapeutic setting, pharmacodynamic relationships of oral cyclodextrin itraconazole have been investigated in a cohort of 27 HIV-infected children with oropharyngeal candidiasis using a standardized scoring of mucosal involvement as endpoint of efficacy and pharmacodynamic modeling [89]. Pharmacokinetic/pharmacodynamic relationships fitted into inhibitory maximum effect pharmacodynamic models. Best fits were observed for AUC, AUC/MIC, Cmax, and Cmax/MIC (r=0.483–0.595, P<0.01), and estimated EC50 values for peak and trough concentrations were 0.40 and 0.16 µg/ml, respectively.
Although several details remain to be investigated, the data presented in this section suggest that, similar to fluconazole, itraconazole exhibits concentration- and time- or exposure-dependent pharmacodynamics (Tables 1 and 2) and that it may be most effective when maintained at certain concentrations at the target site. Minimum trough levels of 0.5 µg/ml (high-performance liquid chromatograph) appear to be a valid surrogate for these concentrations and should be exceeded when itraconazole is given for prophylaxis or treatment of invasive fungal infections. Nevertheless, in the therapeutic situation, the optimum exposure is not well defined since no correlations have been established between pharmacokinetic parameters and measures of efficacy.
Voriconazole
In Vitro Pharmacodynamics
Similar to itraconazole, voriconazole exerts species- and strain-dependent fungicidal activity against opportunistic filamentous fungi, but, on the basis of currently accepted in vitro testing methods, is generally believed to be fungistatic against yeast-like fungi [72, 78, 90]. It is unclear at present whether voriconazole differs from itraconazole, posaconazole, or ravuconazole with respect to its ability to express fungicidal activity.
In vitro, against two strains each of Candida albicans, Candida glabrata, and Cryptococcus neoformans and one strain of Candida tropicalis exposed to concentrations of 0.06–16 times the MIC in RMPI, voriconazole exhibited non-concentration-dependent in vitro pharmacodynamics. Maximum fungistatic activity was noted at drug concentrations of approximately three times the MIC, and neither the EC50 nor the EC90 changed over time [91]. Similar observations of non-concentration-dependent fungistatic activity against Candida albicans have been made by other investigators [72]. In time-kill assays against Aspergillus fumigatus, at concentrations exceeding the MIC for the study isolate by 2.5- to 20-fold, voriconazole displayed time-dependent fungicidal activity [72]. Of note, against Candida albicans, a concentration-dependent PAFE of 0.2–4.1 hours has been observed for voriconazole at concentrations at or four times above MIC in the presence of serum but not in serum-free medium. Similarly, pretreated organisms were more susceptible to subsequent exposure to subinhibitory concentrations than untreated organisms and to killing by polymorphonuclear leukocytes in the presence of serum [92]. It is conceivable that the combination of these effects adds to the in vitro activity of voriconazole and enhances its efficacy against Candida in vivo.
In Vivo Pharmacodynamics and Clinical Implications
The in vivo pharmacodynamics of voriconazole have been investigated in a murine kidney target model of disseminated candidiasis. Similar to other antifungal triazoles, the AUC/MIC ratio was the pharmacodynamic parameter that correlated best with efficacy. Using 10 Candida albicans isolates of different voriconazole susceptibilities, the free drug AUC/MIC ratios were similar for all of the organisms studied (mean±SD, 24±17; P=0.45) and similar to those observed for fluconazole and ravuconazole in the same model [93]. Current dosage recommendations for patients are based on interspecies scaling and exposure-dependent pharmacodynamics such as class effect, dose escalation in non-life-threatening fungal infections, and documented efficacy in the treatment of invasive fungal infections. Since voriconazole has nonlinear pharmacokinetics at therapeutic dosages and exhibits considerable interindividual variability in hepatic metabolization, correlating its pharmacokinetics with antifungal efficacy in patients will be a formidable challenge.
Investigational Triazoles
Investigational antifungal triazoles include posaconazole (SCH 56592; Schering-Plough, Kenilworth, NJ, USA) and ravuconazole (Bristol-Myers Squibb, Wallingford, CT, USA). While ravuconazole is structurally related to fluconazole, the structure of posaconazole is very similar to that of itraconazole. Similar to voriconazole, these new agents have enhanced target activity and specificity. They are active against a wide spectrum of clinically important fungi, including Candida spp., Trichosporon asahii, Cryptococcus neoformans, Aspergillus spp., Fusarium spp., and other hyaline moulds, and against dematiaceous as well as dimorphic moulds [66, 94, 95].
Posaconazole and ravuconazole are highly (>95%) protein bound, exhibit linear pharmacokinetics over the anticipated dosage range, undergo hepatic metabolism, have the potential for significant drug-drug interactions through handling by the CYP450 enzyme system, and are eliminated predominantly via the feces. A hallmark of ravuconazole is its prolonged elimination half-life [96, 97, 98, 99]. Whether the described differences in metabolism and elimination are of clinical significance remains to be determined.
Although posaconazole and ravuconazole follow the general pattern of extended-spectrum triazoles with respect to potential for fungicidal activity against opportunistic moulds and for fungistatic activity against yeasts [79, 100, 101], detailed in vitro pharmacokinetic studies have not been published. The relationships between concentration, time, and antifungal efficacy in vivo have been studied for posaconazole and ravuconazole in animal models of pulmonary aspergillosis and disseminated candidiasis, respectively. The results of these studies are in support of exposure-dependent antifungal efficacy in vivo [102, 103].
IV. Echinocandin Lipopeptides
Class and Mechanism of Action
The echinocandins are a novel class of antifungal lipopeptides. They inhibit the synthesis of 1,3-beta-D-glucan, a polysaccharide in the cell wall of many pathogenic fungi. Together with chitin, the rope-like glucan fibrils are responsible for the cell wall’s strength and shape. They contribute to maintaining the osmotic integrity of the fungal cell and play an important role in cell division and cell growth [104, 105]. While caspofungin (MK-0991; Merck, Rahway, NJ, USA) has been approved in the USA and most of Europe, micafungin (FK463; Fujisawa, Deerfield, IL, USA), and anidulafungin (VER-002, formerly LY303366; Vicuron Pharmaceuticals, Fremont, CA, USA) are in advanced stages of clinical development.
Antifungal Spectrum and Pharmacokinetics
The three current echinocandin agents appear to possess similar pharmacological properties. All three compounds have potent, broad-spectrum fungicidal in vitro activity against Candida spp. and potent fungistatic activity against Aspergillus spp.; their antifungal efficacy against these organisms has been demonstrated in various animal models and in patients. The echinocandins are not metabolized through the CYP450 enzyme system and are generally well tolerated due to the lack of mechanism-based toxicity. Although only available in parenteral formulations, the echinocandins possess favorable pharmacokinetic properties and are dosed once daily [106, 107].
Pharmacodynamics In Vitro
The current echinocandins possess a species-dependent mode of antifungal activity. Whereas potent inhibition of glucan synthesis has been demonstrated in membrane preparations of Candida and Aspergillus spp., whole cell in vitro assays reveal fungicidal activity against most Candida spp. but not against Aspergillus spp. [108, 109]. Broth-based assays show a dose-dependent formation of microcolonies [110, 111] with progressively truncated, swollen hyphal elements that appear to be cell-wall deficient but regain their cell walls following withdrawal of the drug [110, 111, 112]. Vitality fluorescence staining of Aspergillus fumigatus exposed to caspofungin revealed a differential killing effect on apical and subapical branching cells, with little reduction in vital staining of subapical cells, suggesting that only cells at the active centers for new cell wall synthesis within Aspergillus fumigatus hyphae are killed [113]. These differences may have implications for antifungal therapy of the neutropenic host. In infection models in persistently neutropenic rabbits, echinocandins achieved a dose-dependent elimination of Candida spp. in experimental disseminated candidiasis [114, 115]; however, they had no effect on the residual fungal burden in experimental pulmonary aspergillosis and were less effective than amphotericin B in attenuating tissue injury and improving survival [115, 116, 117].
In vitro time-kill assays have demonstrated concentration-dependent fungicidal activity (≥99.9% reduction in cfu) against Candida spp. for caspofungin [118], micafungin [115], and anidulafungin [114, 119, 120]. In addition, a concentration-dependent PAFE exceeding 12 hours has been observed for both caspofungin and anidulafungin against Candida spp. [47]. In a one-compartment in vitro pharmacodynamic model of anidulafungin against fluconazole-sensitive and fluconazole-resistant Candida albicans, a positive correlation of Cmax and AUC with optimal killing and minimal fungal regrowth has been demonstrated [121].
Pharmacodynamics In Vivo
In vivo, in persistently neutropenic rabbit models of subacute disseminated candidiasis and invasive pulmonary aspergillosis, anidulafungin showed highly predictable concentration-effect relationships in experimental disseminated candidiasis; however, no concentration-effect relationships were observed in experimental pulmonary aspergillosis using the residual fungal burden and survival as endpoints of antifungal efficacy, despite full exploration of the dosage range [122]. Similar concentration-dependent activity against Candida albicans was found in a thigh infection model: the ratio between tissue concentrations and MIC was found to be highly predictive of the therapeutic efficacy of micafungin[123]. In murine kidney target models of disseminated candidiasis, the AUC/MIC ratio was most strongly correlated with efficacy of caspofungin [124], whereas the peak/MIC ratio was most predictive of the efficacy of HMR 3270, a novel investigational glucan synthesis inhibitor [125].
Clinical Implications
Concentration-dependent pharmacodynamics in vitro and in vivo (Tables 1 and 2) and the existence of prolonged PAFEs support the current once-daily dosing regimen and, at the same time, provide a rationale for exploration of dose escalation for treatment of complicated invasive Candida infections. The pharmacodynamics of echinocandins against Aspergillus spp. are more complex and difficult to interpret, and perhaps suggest concentration-independent dynamics beyond a certain threshold dosage.
Pharmacodynamics of Antifungal Combinations
The availability of drugs with different molecular targets has opened new avenues for exploring combination therapies of two or even three drugs. The obvious aims of combination therapies are to broaden the antifungal spectrum, to decrease the selection of resistant organisms, and to improve overall antifungal efficacy without compromising patient safety. However, combination therapy is not to be perceived as the universal approach for all proven or suspected fungal infections but will be reserved for selected subgroups. These include patients with invasive infections due to opportunistic filamentous fungi and poor prognosis, those with acute and fulminant or refractory infections, and those with infections at anatomically privileged sites such as the brain [126, 127].
The paradigm for this approach is the combination of amphotericin B deoxycholate and flucytosine, which has documented synergistic activity against Cryptococcus neoformans in vitro and in animal models [128], activity that also translated into superior outcome in patients with cryptococcal meningoencephalitis [129, 130]. Beyond cryptococcal meningitis, clinical experience with combination therapies is mostly anecdotal. However, there is an understandable trend to utilize in desperately ill patients whatever combination appears to have a theoretical advantage. Nevertheless, irrespective of the pressing clinical need, systematic preclinical investigation of antifungal combination therapies is warranted, followed by appropriately designed randomized clinical trials. This may be illustrated by a drug- and fungus-specific antagonism between amphotericin B deoxycholate and antifungal azoles in vitro and in animal models that has been consistently noted with lipophilic azoles against Candida spp. and, in particular, against Aspergillus spp. [131, 132, 133]. A recently completed placebo-controlled study comparing fluconazole 800 mg/day plus placebo versus fluconazole 800 mg/day plus amphotericin B (0.7 mg/kg/day) for treatment of non-neutropenic candidemia, however, revealed no evidence of antagonism but more rapid clearance of the bloodstream and a trend toward improved antifungal efficacy of the combination [134]. While the combination of amphotericin B and itraconazole continues to be controversial [126, 127], the body of current preclinical studies indicate no antagonism of combinations of echinocandins with azoles, amphotericin B, or the chitin-synthase inhibitor nikkomycin Z against common opportunistic fungal pathogens [135, 136]. In fact, synergistic efficacy has been observed for the combination of echinocandins and novel triazoles against experimental invasive aspergillosis [137, 138], and first preliminary reports are emerging that point to the potential clinical usefulness of combination therapies in the treatment of invasive aspergillosis [139, 140].
Over the next decade, combination therapy will probably become the standard of care for fungal infections that are notoriously difficult to treat. However, the critical question of how these combinations can be harnessed to improve antifungal therapy can only be evaluated in sufficiently powered, randomized clinical trials that are founded on discriminative animal infection models that incorporate pharmacokinetic and pharmacodynamic endpoints [141].
Future Directions
Invasive fungal infections will likely remain a frequent and important complication in the ever-expanding population of immunocompromised patients. However, not unlike the current situation with antibacterial agents, the road to identification of novel targets and the development of entirely new antifungal therapeutics is difficult and paved with failed projects. One reason for the slow progress in this area is that, owing to their eukaryotic nature, fungal cells have a restricted set of specific molecular targets that do not overlap with their mammalian counterparts and carry the potential of mechanism-based toxicity. In the long range, whole-genome sequencing, bioinformatics, and advances in proteomics and stereochemistry hold great promise for accelerated identification and development of selective antifungal compounds [142]. Nevertheless, for the next foreseeable future, our antifungal armamentarium is likely to remain limited. For this reason, continuing efforts are needed to further capitalize on the current experimental and clinical foundations of antifungal drug research and therapy. The now considerably expanded antifungal drug arsenal, predictive resistance testing in vitro, integration of pharmacokinetic and pharmacodynamic principles, and multiple-drug regimens offer hope for further substantial progress in prevention and treatment of invasive fungal infections.
References
Groll AH, Walsh TJ (2001) Uncommon opportunistic fungi: new nosocomial threats. Clin Microbiol Infect 7 (Suppl 2):8–24
Schneider E, Hajjeh RA, Spiegel RA, Jibson RW, Harp EL, Marshall GA, Gunn RA, McNeil MM, Pinner RW, Baron RC, Burger RC, Hutwagner LC, Crump C, Kaufman L, Reef SE, Feldman GM, Pappagianis D, Werner SB (1997) A coccidioidomycosis outbreak following the Northridge, Calif., earthquake. JAMA 277:904–908
Centers for Disease Control (2001) Update: outbreak of acute febrile respiratory illness among college students – Acapulco, Mexico, March 2001. Morb Mortal Wkly Rep 50:359–360
Groll, AH, Piscitelli SC, Walsh TJ (1998) Clinical pharmacology of systemic antifungal agents: a comprehensive review of agents in clinical use, current investigational compounds, and putative targets for antifungal drug development. Adv Pharmacol 44:343–500
Berenguer J, Buck M, Witebsky F, Stock F, Pizzo PA, Walsh TJ (1993) Lysis-centrifugation blood cultures in the detection of tissue-proven invasive candidiasis. Disseminated versus single-organ infection. Diagn Microbiol Infect Dis 17:103–109
Hebart H, Loffler J, Meisner C et al (2000) Early detection of Aspergillus infection after allogeneic stem cell transplantation by polymerase chain reaction screening. J Infect Dis 181:1713–1719
Maertens J, Verhaegen J, Lagrou K et al (2001) Screening for circulating galactomannan as a noninvasive diagnostic tool for invasive aspergillosis in prolonged neutropenic patients and stem cell transplantation recipients: a prospective validation. Blood 97:1604–1610
Caillot D, Casasnovas O, Bernard A et al (1997) Improved management of invasive pulmonary aspergillosis in neutropenic patients using early thoracic computed tomography scan and surgery. J Clin Oncol 15:139–147
Caillot D, Couaillier JF, Bernard A, Casasnovas O, Denning DW, Mannone L, Lopez J, Couillault G, Piard F, Vagner O, Guy H (2001) Increasing volume and changing characteristics of invasive pulmonary aspergillosis on sequential thoracic computed tomography scans in patients with neutropenia. J Clin Oncol 19:253–259
Walsh TJ, Roden M, Roilides E, Groll AH (2000) Concepts in design of comparative clinical trials of antifungal therapy in neutropenic patients. Int J Antimicrob Agents 16:151–156
Rex JR, Walsh TJ, Nettleman M et al (2001) Need for alternative trial designs and evaluation strategies for therapeutic studies of invasive mycoses. Clin Infect Dis 35:95–106
Ascioglu S, Rex JH, de Pauw B, Bennett JE, Bille J, Crokaert F et al (2002) Defining opportunistic invasive fungal infections in immunocompromised patients with cancer and hematopoietic stem cell transplants: an international consensus. Clin Infect Dis 34:7–14
Rex JH, Pfaller MA, Galgiani JN, Bartlett MS, Espinel-Ingroff A, Ghannoum MA, Lancaster M, Odds FC, Rinaldi MG, Walsh TJ, Barry AL (1997) Development of interpretive breakpoints for antifungal susceptibility testing: conceptual framework and analysis of in vitro-in vivo correlation data for fluconazole, itraconazole, and Candida infections. Clin Infect Dis 24:235–247
Groll AH, Piscitelli SC, Walsh TJ (2001) Antifungal pharmacodynamics: concentration-effect relationships in vitro and in vivo. Pharmacotherapy 21 (Suppl):133–148
Andes D (2003) In vivo pharmacodynamics of antifungal drugs in treatment of candidiasis. Antimicrob Agents Chemother 47:1179–1186
Woods GL (1995) In vitro testing of antimicrobial agents. Infect Dis Clin North Am 9:463–481
National Committee for Clinical Laboratory Standards (1997) Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard M27-A. NCCLS, Wayne, PA
Cuenca-Estrella M, Lee-Yang W, Ciblak MA, Arthington-Skaggs BA, Mellado E, Warnock DW, Rodriguez-Tudela JL (2002) Comparative evaluation of NCCLS M27-A and EUCAST broth microdilution procedures for antifungal susceptibility testing of Candida species. Antimicrob Agents Chemother 46:3644–3647
National Committee for Clinical Laboratory Standards (2000) Reference method for broth dilution antifungal susceptibility testing of conidium-forming filamentous fungi. Approved standard M38-A. NCCLS, Wayne, PA
Rex JH, Pfaller MA (2002) Has antifungal susceptibility testing come of age? Clin Infect Dis 35:982–989
Perea S, Patterson TF (2002). Antifungal resistance in pathogenic fungi. Clin Infect Dis 35:1073–1080
Vanden Bossche H, Dromer F, Improvisi I, Lozano-Chiu M, Rex JH, Sanglard D (1998) Antifungal drug resistance in pathogenic fungi. Med Mycol 36 (Suppl 1):119–128
Lopez-Ribot JL, McAtee RK, Lee LN, Kirkpatrick WR, White TC, Sanglard D, Patterson TF (1998) Distinct patterns of gene expression associated with development of fluconazole resistance in serial Candida albicans isolates from human immunodeficiency virus-infected patients with oropharyngeal candidiasis. Antimicrob Agents Chemother 42:2932–2937
White TC, Marr KA, Bowden RA (1998) Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clin Microbiol Rev 11:382–402
Dannaoui E, Borel E, Monier MF, Piens MA, Picot S, Persat F (2001) Acquired itraconazole resistance in Aspergillus fumigatus. J Antimicrob Chemother 47:333–340
Hof H (2001) Critical annotations to the use of azole antifungals for plant protection. Antimicrob Agents Chemother 45:2987–2990
Vanden Bossche H, Engelen M, Rochette F. (2003) Antifungal agents of use in animal health – chemical, biochemical and pharmacological aspects. J Vet Pharmacol Ther 26:5–29
Morschhauser J (2002) The genetic basis of fluconazole resistance development in Candida albicans. Biochim Biophys Acta 1587:240–248
Sanglard D, Odds FC (2002) Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect Dis 2:73–85
Kontoyiannis DP, Lewis RE (2002) Antifungal drug resistance of pathogenic fungi. Lancet 359:1135–1144
Kurtz MB, Abruzzo G, Flattery A, Bartizal K, Marrinan JA, Li W, Milligan J, Nollstadt K, Douglas CM (1996) Characterization of echinocandin-resistant mutants of Candida albicans: genetic, biochemical, and virulence studies. Infect Immun 64:3244–3251
Craig WA (1998) Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 26:1–10
Janknegt R, de Marie S, Bakker-Woudenberg IA, Crommelin DJ (1992) Liposomal and lipid formulations of amphotericin B. Clinical pharmacokinetics. Clin Pharmacokinet 23:279–291
Hiemenz JW, Walsh TJ (1996) Lipid formulations of amphotericin B: recent progress and future directions. Clin Infect Dis 22 (Suppl 2):133–144
Wong-Beringer A, Jacobs RA, Guglielmo BJ (1998) Lipid formulations of amphotericin B: clinical efficacy and toxicities. Clin Infect Dis 27:603–618
Chiou C, Groll AH, Walsh TJ (2000) New drugs and novel targets for treatment of invasive fungal infections in patients with cancer. Oncologist 5:120–135
Brajtburg J, Powderly WG, Kobayashi GS, Medoff G (1999) Amphotericin B: current understanding of mechanisms of action. Antimicrob Agents Chemother 34:183–188
Bekersky I, Fielding RM, Dressler DE, Lee JW, Buell DN, Walsh TJ (2002) Plasma protein binding of amphotericin B and pharmacokinetics of bound versus unbound amphotericin B after administration of intravenous liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate. Antimicrob Agents Chemother 46:834–840
Bekersky I, Fielding RM, Dressler DE, Lee JW, Buell DN, Walsh TJ (2002) Pharmacokinetics, excretion, and mass balance of liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate in humans. Antimicrob Agents Chemother 46:828–833
Bekersky I, Fielding RM, Dressler DE, Kline S, Buell DN, Walsh TJ (2001) Pharmacokinetics, excretion, and mass balance of 14C after administration of 14C-cholesterol-labeled AmBisome to healthy volunteers. J Clin Pharmacol 41:963–971
Klepser ME, Wolfe EJ, Jones RN, et al (1997) Antifungal pharmacodynamic characteristics of fluconazole and amphotericin B tested against Candida albicans. Antimicrob Agents Chemother 41:1392–1395
Klepser ME, Wolfe EJ, Pfaller MA (1998) Antifungal pharmacodynamic characteristics of fluconazole and amphotericin B against Cryptococcus neoformans. J Antimicrob Chemother 41:397–401
Walsh TJ (1989) Trichosporonosis. Infect Dis Clin North Amer 3:43–52
Walsh TJ, Melcher G, Rinaldi M, Lecciones J, McGough D, Lee J, Callender D, Rubin M, Pizzo PA (1990) Trichosporon beigelii: an emerging pathogen resistant to amphotericin B. J Clin Microbiol 28:1616–1622
Walsh TJ, Lee JW, Melcher GP, Navarro E, Bacher J, Callender D, Reed KD, Wu T, Lopez-Berestein G, Pizzo PA (1992) Experimental disseminated trichosporonosis in persistently granulocytopenic rabbits: implications for pathogenesis, diagnosis, and treatment of an emerging opportunistic infection. J Infect Dis 166:121–133
Turnidge JD, Gudmundsson S, Vogelman B, Craig WA (1994) The postantibiotic effect of antifungal agents against common pathogenic yeasts. J Antimicrob Chemother 34:83–92
Ernst E, Klepser ME, Pfaller MA (2000) Postantifungal effects of echinocandin, azole, and polyene antifungal agents Candida albicans and Cryptococcus neoformans. Antimicrob Agents Chemother 44:1108–1111
Andes D, Stamsted T, Conklin R (2001) Pharmacodynamics of amphotericin B in a neutropenic-mouse disseminated-candidiasis model. Antimicrob Agents Chemother 45:922–926
Hoffman HL, Lewis RE, Ernst EJ, et al (2000) In vivo pharmacodynamics of liposomal amphotericin B against Candida albicans in a neutropenic murine lung infection model. Pharmacotherapy 20:357–358
Groll AH, Giri N, Petraitis V, et al\ (2000) Comparative efficacy and distribution of lipid formulations of amphotericin B in experimental Candida albicans infection of the central nervous system. J Infect Dis 182:274–282
Walsh TJ, Goodman JL, Pappas P, Bekersky I, Buell DN, Roden M, Barrett J, Anaissie EJ (2001) Safety, tolerance, and pharmacokinetics of high-dose liposomal amphotericin B (AmBisome) in patients infected with Aspergillus species and other filamentous fungi: maximum tolerated dose study. Antimicrob Agents Chemother 45:3487–3496
Walsh TJ, Jackson AJ, Lee JW, Amantea M, Sein T, Bacher J, Zech L (2000) Dose-dependent pharmacokinetics of amphotericin B lipid complex in rabbits. Antimicrob Agents Chemother 44:2068–2076
Walsh TJ, Whitcomb T, Piscitelli S, Figg WD, Hill S, Chanock SJ, Jarosinski P, Pizzo PA (1997) Safety, tolerance, and pharmacokinetics of amphotericin B lipid complex in children with hepatosplenic candidiasis. Antimicrob Agents Chemother 41:1944–1948
Polak A, Scholer HJ (1975) Mode of action of 5-fluorocytosine and mechanisms of resistance. Chemotherapy 21:113–130
Pfaller MA, Messer SA, Coffman S (1997) In vitro susceptibilities of clinical yeast isolates to a new echinocandin derivative, LY303366, and other antifungal agents. Antimicrob Agents Chemother 41:763–766
Hoban DJ, Zhanel GG, Karlowsky JA (1999) In vitro susceptibilities of Candida and Cryptococcus neoformans isolates from blood cultures of neutropenic patients. Antimicrob Agents Chemother 43:1463–1464
Vermes A, Guchelaar HJ, Dankert J (2000) Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J Antimicrob Chemother 46:171–179
Brandt ME, Pfaller MA, Hajjeh RA, Hamill RJ, Pappas PG, Reingold AL, Rimland D, Warnock DW (2001) Cryptococcal Disease Active Surveillance Group. Trends in antifungal drug susceptibility of Cryptococcus neoformans isolates in the United States: 1992 to 1994 and 1996 to 1998. Antimicrob Agents Chemother 45:3065–3069
Francis P, Walsh TJ (1992) Evolving role of flucytosine in immunocompromised patients: new insights into safety, pharmacokinetics, and antifungal therapy. Clin Infect Dis 15:1003–1018
van der Auwera P, Ceuppens AM, Heymans C, Meunier F (1986) In vitro evaluation of various antifungal agents alone and in combination by using an automatic turbidimetric system combined with viable count determinations. Antimicrob Agents Chemother 29:997–1004
Lewis RE, Klepser ME, Pfaller MA (2000) In vitro pharmacodynamic characteristics of flucytosine determined by time-kill methods. Diagn Microbiol Infect Dis 36:101–105
Scalarone GM, Mikami Y, Kurita N, Yazawa K, Miyaji M (1992) The postantifungal effect of 5-fluorocytosine on Candida albicans. J Antimicrob Chemother 29:129–136
Andes D, van Ogtrop M (2000) In vivo characterization of the pharmacodynamics of flucytosine in a neutropenic murine disseminated candidiasis model. Antimicrob Agents Chemother 44:938–942
Grant SM, Clissold SP (1990) Fluconazole. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in superficial and systemic mycoses. Drugs 39:877–916
Grant SM, Clissold SP (1989) Itraconazole. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in superficial and systemic mycoses. Drugs 37:310–344
Groll AH, Gea-Banacloche JC, Glasmacher A, Just-Nuebling G, Maschmeyer G, Walsh TJ (2003) Clinical pharmacology of antifungal compounds. Infect Dis Clin North Am 17:159–191
Anaissie EJ, Kontoyiannis DP, Huls C, Vartivarian SE, Karl C, Prince RA, Bosso J, Bodey GP (1995) Safety, plasma concentrations, and efficacy of high-dose fluconazole in invasive mold infections. J Infect Dis 172:599–602
Hoffman HL, Ernst EJ, Klepser ME (2000) Novel triazole antifungal agents. Expert Opin Investig Drugs 9:593–605
Sohnle PG, Hahn BL, Erdmann MD (1996) Effect of fluconazole on viability of Candida albicans over extended periods of time. Antimicrob Agents Chemother 40:2622–2625
Walsh TJ, Aoki S, Mechinaud F, Bacher J, Lee J, Rubin M, Pizzo PA (1990) Effects of preventive, early, and late antifungal chemotherapy with fluconazole in different granulocytopenic models of experimental disseminated candidiasis. J Infect Dis 161:755–760
Anaissie EJ, Darouiche RO, Abi-Said D et al (1996) Management of invasive Candida infections: results of a prospective, randomized, multicenter study of fluconazole versus amphotericin B and review of the literature. Clin Infect Dis 23:964–972
Anaissie EJ, Vartivarian SE, Abi-Said D et al (1996) Fluconazole versus amphotericin B in the treatment of hematogenous candidiasis: a matched cohort study. Am J Med 101:170–176
Minguez F, Chiu ML, Lima JE, Nique R, Prieto J (1994) Activity of fluconazole: postantifungal effect, effects of low concentrations and of pretreatment on the susceptibility of Candida albicans to leucocytes. J Antimicrob Chemother 34:93–100
Lewis RE, Lund BC, Klepser ME, Ernst EJ, Pfaller MA (1998) Assessment of antifungal activities of fluconazole and amphotericin B administered alone and in combination against Candida albicans by using a dynamic in vitro mycotic infection model. Antimicrob Agents Chemother 42:1382–1386
Louie A, Drusano GL, Banerjee P, Liu QF, Liu W, Kaw P, Shayegani M, Taber H, Miller MH (1998) Pharmacodynamics of fluconazole in a murine model of systemic candidiasis. Antimicrob Agents Chemother 42:1105–1109
Andes D, van Ogtrop M (1999) Characterization and quantitation of the pharmacodynamics of fluconazole in a neutropenic murine disseminated candidiasis infection model. Antimicrob Agents Chemother 43:2116–2120
Manavathu EK, Cutright JL, Chandrasekar PH (1998) Organism-dependent fungicidal activities of azoles. Antimicrob Agents Chemother 42:3018–3021
Johnson EM, Szekely A, Warnock DW (1998) In vitro activity of voriconazole, itraconazole and amphotericin B against filamentous fungi. J Antimicrob Chemother 42:741–745
Fung-Tomc JC, White TC, Minassian B, Huczko E, Bonner DP (1999) In vitro antifungal activity of BMS-207147 and itraconazole against yeast strains that are non-susceptible to fluconazole. Diagn Microbiol Infect Dis 35:163–167
Burgess DS, Hastings RW (2000) A comparison of dynamic characteristics of fluconazole, itraconazole, and amphotericin B against Cryptococcus neoformans using time-kill methodology. Diagn Microbiol Infect Dis 38:87–93
Burgess DS, Hastings RW, Summers KK, Hardin TC, Rinaldi MG (2000) Pharmacodynamics of fluconazole, itraconazole, and amphotericin B against Candida albicans. Diagn Microbiol Infect Dis 36:13–18
Zhanel GG, Saunders DG, Hoban DJ, Karlowsky JA (1999) Amphotericin B, azole, and 5-flucytosine pharmacodynamic parameters in the presence of human serum. In: Program and abstracts of the 39th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, p 23
Denning DW, Radford SA, Oakley KL, Hall L, Johnson EM, Warnock DW (1997) Correlation between in vitro susceptibility testing to itraconazole and in vivo outcome of Aspergillus fumigatus infection. J Antimicrob Chemother 40:401–414
Berenguer J, Ali NM, Allende MC, Lee J, Garrett K, Battaglia S, Piscitelli SC, Rinaldi MG, Pizzo PA, Walsh TJ (1994) Itraconazole for experimental pulmonary aspergillosis: comparison with amphotericin B, interaction with cyclosporine A, and correlation between therapeutic response and itraconazole concentrations in plasma. Antimicrob Agents Chemother 38:1303–1308
Groll AH, Mickiene D, Petraitiene R, Petraitis V, Roussillion K, Hemmings M, Raskas S, Walsh TJ (2001) Dose escalation pharmacodynamic study of intravenous itraconazole in a neutropenic animal model of invasive pulmonary aspergillosis. In: Program and abstracts of the 41st Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, p 389
Boogaerts MA, Verhoef GE, Zachee P, Demuynck H, Verbist L, De Beule K (1989) Antifungal prophylaxis with itraconazole in prolonged neutropenia: correlation with plasma levels. Mycoses 32 (Suppl 1):103–108
De Beule K (1996) Itraconazole: pharmacology, clinical experience and future development. Int J Antimicrobial Agents 6:175–181
Glasmacher A, Hahn C, Molitor E, Sauerbruch T, Marklein G, Schmidt-Wolf IGH (2000) Definition of itraconazole target concentration for antifungal prophylaxis. In: Programs and abstracts of the 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, p 363
Groll AH, Mickiene D, McEvoy M, Dad L, Townley E, Piscitelli S, Wood L, Walsh TJ (2002) Safety, pharmacokinetics and pharmacodynamics of cyclodextrin itraconazole in pediatric patients with oropharyngeal candidiasis. Antimicrob Agents Chemother 46:2554–2563
Clancy CJ, Nguyen MH (1998) In vitro efficacy and fungicidal activity of voriconazole against Aspergillus and Fusarium species. Eur J Clin Microbiol Infect Dis 17:573–575
Klepser ME, Malone D, Lewis RE, Ernst EJ, Pfaller MA (2000) Evaluation of voriconazole pharmacodynamics using time-kill methodology. Antimicrob Agents Chemother 44:1917–1920
Garcia MT, Llorente MT, Lima JE, Minguez F, Del Moral F, Prieto J (1999) Activity of voriconazole: post-antifungal effect, effects of low concentrations and of pretreatment on the susceptibility of Candida albicans to leucocytes. Scand J Infect Dis 31:501–504
Andes D, Marchillo K, Stamstad T, Conklin R (2003) In vivo pharmacokinetics and pharmacodynamics of a new triazole, voriconazole, in a murine candidiasis model. Antimicrob Agents Chemother 47:3165–3169
Pfaller MA, Messer SA, Hollis RJ, Jones RN, Doern GV, Brandt ME, Hajjeh RA (1998) In vitro susceptibilities of Candida bloodstream isolates to the new triazole antifungal agents BMS-207147, Sch 56592, and voriconazole. Antimicrob Agents Chemother 42:3242–3244
Pfaller MA, Messer SA, Hollis RJ, Jones RN, and the SENTRY Participants Group (2002) Antifungal activities of posaconazole, ravuconazole, and voriconazole compared to those of itraconazole and amphotericin B against 239 clinical isolates of Aspergillus species and other filamentous fungi: report from SENTRY Antimicrobial Surveillance Program, 2000. Antimicrob Agents Chemother 46:1032–1037
Krieter P, Flannery B, Musick T, Courtney R, Patrick J, Laughlin M (2002) Pharmacokinetics and excretion of 14c posaconazole following oral administration in healthy male subjects. In: Program and abstracts of the 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, p 20
Wexler D, Laughlin M, Lim J, Courtney R, Batra V (2002) Effect of posaconazole on drug metabolizing enzymes. In: Program and abstracts of the 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, p 30
Olsen SJ, Mummaneni V, Rolan P, Norton J, Grasela DM (2000) Ravuconazole: single ascending oral dose study in healthy subjects. In: Program and abstracts of the 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, p 22
Grasela DM, Olsen SJ, Rolan P, Mummaneni V, Birkhofer MJ, Christopher L, Norton J, Grasela DM, Hadjilambris OH, Marino MR (2000) Ravuconazole: multiple ascending oral dose study in healthy subjects. In: Program and abstracts of the 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, p 22
Fung-Tomc JC, Huczko E, Minassian B, Bonner DP (1998) In vitro activity of a new oral triazole, BMS-207147 (ER-30346). Antimicrob Agents Chemother 42:313–318
Espinel-Ingroff A (1998) Comparison of in vitro activities of the new triazole SCH56592 and the echinocandins MK-0991 (L-743,872) and LY303366 against opportunistic filamentous and dimorphic fungi and yeasts. J Clin Microbiol 36:2950–2956
Groll AH, Mickiene D, Petraitiene R, Petraitis V, Sein T, Piscitelli SC, Walsh TJ. (2000) Pharmacokinetics and pharmacodynamics of posaconazole (SCH 56592) in a neutropenic animal model of invasive pulmonary aspergillosis. In: Program and abstracts of the 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, p 385
Andes D, Marchillo K, Stamstad T, Conklin R (2003) In vivo pharmacodynamics of a new triazole, ravuconazole, in a murine candidiasis model. Antimicrob Agents Chemother 47:1193–1199
Hector RF (1993) Compounds active against cell walls of medically important fungi. Clin Microbiol Rev 6:1–21
Debono M, Gordee RS (1994) Antibiotics that inhibit fungal cell wall development. Ann Rev Microbiol 48:471–497
Denning DW (2002) Echinocandins: a new class of antifungal agents. J Antimicrob Chemother 49:889–891
Georgopapadakou NH (2001) Update on antifungals targeted to the cell wall: focus on beta-1,3-glucan synthase inhibitors. Expert Opin Investig Drugs 10:269–280
Tang J, Parr TR, Turner W, Debono M, Lagrandeur L, Burkhard F, Rodriguez M, Zweifel M, Nissen J, Clingerman K (1993) LY-303366: a non-competitive inhibitor of (1,3)-b-D glucan synthases from Candida albicans and Aspergillus fumigatus. In: Program and abstracts of the 33rd Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, Abstract no. 367
Maki K, Morishita Y, Iguchi Y, Watabe E, Otomo K, Teratani M, Watanabe Y, Ikeda F, Tawara S, Goto T, Tomishima M (1998) In vitro antifungal activity of FK463, a novel water-soluble echinocandin-like lipopeptide. In: Program and abstracts of the 38th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, Abstract no. F141
Kurtz MB, Heath IB, Marrinan J, Dreikorn S, Onishi J, Douglas C (1994) Morphological effects of lipopeptides against Aspergillus fumigatus correlate with activities against (1,3)-beta-D-glucan synthase. Antimicrob Agents Chemother 38:1480–1489
Oakley KL, Moore CB, Denning DW (1998) In vitro activity of the echinocandin antifungal agent LY303,366 in comparison with itraconazole and amphotericin B against Aspergillus species. Antimicrob Agents Chemother 42:2726–2730
Rennie R, Sand C, Sherburne R (1997) Electron microscopic evidence of the effect of LY303366 on Aspergillus fumigatus. In: Abstracts of the 13th Meeting of the International Society for Human and Animal Mycology, Abstract no. P451
Bowman JC, Hicks PS, Kurtz MB, Rosen H, Schmatz DM, Liberator PA, Douglas CM (2002) The antifungal echinocandin caspofungin acetate kills growing cells of Aspergillus fumigatus in vitro. Antimicrob Agents Chemother 46:3001–3012
Petraitiene R, Petraitis V, Groll AH, Candelario M, Sein T, Bell A, Lyman CA, McMillian CL, Bacher J, Walsh TJ (1999) Antifungal activity of LY303366, a novel echinocandin B, in experimental disseminated candidiasis in rabbits. Antimicrob Agents Chemother 43:2148–2155
Petraitis V, Petraitiene R, Groll AH, Roussillon K, Hemmings M, Lyman CA, Sein T, Bacher J, Bekersky I, Walsh TJ (2002) Comparative antifungal activities and plasma pharmacokinetics of micafungin (FK463) against disseminated candidiasis and invasive pulmonary aspergillosis in persistently neutropenic rabbits. Antimicrob Agents Chemother 46:1857–1869
Petraitis V, Petraitiene R, Groll AH, Bell A, Callender DP, Sein T, Schaufele RL, McMillian CL, Bacher J, Walsh TJ (1998) Antifungal efficacy, safety, and single-dose pharmacokinetics of LY303366, a novel echinocandin B, in experimental pulmonary aspergillosis in persistently neutropenic rabbits. Antimicrob Agents Chemother 42:2898–2905
Petraitiene R, Petraitis V, Groll AH, Sein T, Schaufele RL, Francesconi A, Bacher J, Avila NA, Walsh TJ (2002) Antifungal efficacy of caspofungin (MK-0991) in experimental pulmonary aspergillosis in persistently neutropenic rabbits: pharmacokinetics, drug disposition, and relationship to galactomannan antigenemia. Antimicrob Agents Chemother 46:12–23
Ernst EJ, Klepser ME, Ernst ME, Messer SA, Pfaller MA (1999) In vitro pharmacodynamic properties of MK-0991 determined by time-kill methods. Diagn Microbiol Infect Dis 33:75–80
Klepser ME, Ernst EJ, Ernst ME, Pfaller MA (1997) Growth medium effect on the antifungal activity of LY303366. Diagn Microbiol Infect Dis 29:227–231
Green LJ, Marder P, Mann LL, Chio LC, Current WL (1999) LY303366 exhibits rapid and potent fungicidal activity in flow cytometric assays of yeast viability. Antimicrob Agents Chemother 43:830–835
Zhanel G, Zelenitsky S, Laing N, Balko T, Karlowsky J, Hoban D (1998) Correlation between LY303366 area under the concentration curve (AUC) and regrowth of fluconazole sensitive (flu-s) and fluconazole-resistant (flu-r) Candida albicans, using an in vitro pharmacodynamic model. In: Program and abstracts of the 38th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, Abstract no. J-14
Groll AH, Mickiene D, Petraitiene R, Petraitis V, Lyman CA, Bacher JS, Piscitelli SC, Walsh TJ (2001) Pharmacokinetic and pharmacodynamic modeling of anidulafungin (LY303366): reappraisal of its efficacy in neutropenic animal models of opportunistic mycoses using optimal plasma sampling. Antimicrob Agents Chemother 45:2845–2855
Matsumoto S, Warabe E, Wakai Y, Koide Y, Ushitani T, Teratani N, Ohtomo K, Hatano K, Ikeda F, Goto T, Matsumoto F, Kuwahara S (2000) Pharmacodynamics of FK463 in a thigh infection model with Candida albicans. In: Program and abstracts of the 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, Abstract no. 1687
Louie A, Deziel M, Liu W, Drusano M, Gumbo T, Drusano GL (2003) AUC/MIC is the pharmacodynamic variable for caspofungin as determined in a non-neutropenic mouse model of candidiasis. In: Program and abstracts of the 43rd International Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington DC, Abstract no. A-1572
Andes D, Marchillo K, Lowther J, Bryskier A, Stamstad T, Conklin R (2003) In vivo pharmacodynamics of HMR 3270, a glucan synthase inhibitor, in a murine candidiasis model. Antimicrob Agents Chemother 47:1187–1192
Lewis RE, Kontoyiannis DP (2001) Rationale for combination antifungal therapy. Pharmacotherapy 21 (Suppl):149–164
Sugar AM (2001) Antifungal combination therapy. Curr Opin Invest Drugs 2:1364–1365
Medoff G, Comfort M, Kobayashi GS (1971) Synergistic action of amphotericin B and 5-fluorocytosine against yeast-like organisms. Proc Soc Exp Biol Med 138:571–574
Bennett JE, Dismukes WE, Haywood M, Duma R, Medoff G (1979) A comparison of amphotericin B alone and in combination with flucytosine in the treatment of cryptococcal meningitis. N Engl J Med 301:126-131
van der Horst CM, Saag MS, Cloud GA, et al (1997) Treatment of cryptococcal meningitis associated with the acquired immunodeficiency syndrome. N Engl J Med 337:15–21
Schaffner A, Bohler A (1993) Amphotericin B refractory aspergillosis after itraconazole: evidence for significant antagonism. Mycoses 36:421–424
Sugar AM, Hitchcock CA, Troke PF, Picard M (1995) Combination therapy of murine invasive candidiasis with fluconazole and amphotericin B. Antimicrob Agents Chemother 39:598–601
Sugar AM, Liu XP (1998) Interactions of itraconazole with amphotericin B in the treatment of murine invasive candidiasis. J Infect Dis 177:1660–1663
Rex JH, Pappas PG, Karchmer AW, Sobel J, Edwards JE, Hadley S, Brass C, Vazquez JA, et al, the National Institute of Allergy and Infectious Diseases Mycoses Study Group (2003) A randomized and blinded multicenter trial of high-dose fluconazole plus placebo versus fluconazole plus amphotericin B as therapy for candidemia and its consequences in nonneutropenic subjects. Clin Infect Dis 36:1221–1228
Groll AH, Walsh TJ (2001) Caspofungin: pharmacology, safety and therapeutic potential in superficial and invasive fungal infections. Expert Opin Invest Drugs 10:1545–1558
Groll AH, Walsh TJ (2000) FK-463. Curr Opin Anti-Infect Invest Drugs 2:405–412
Kirkpatrick WR, Perea S, Coco BJ, Patterson TF (2002) Efficacy of caspofungin alone and in combination with voriconazole in a guinea pig model of invasive aspergillosis. Antimicrob Agents Chemother 46:2564–2568
Petraitis V, Petraitiene R, Sarafandi AA, Kelaher AM, Lyman CA, Casler HE, Sein T, Groll AH, Bacher J, Avila NA, Walsh TJ (2003) Combination therapy in treatment of experimental pulmonary aspergillosis: synergistic interaction between an antifungal triazole and an echinocandin. J Infect Dis 187:1834–1843
Rubin MA, Carroll KC, Cahill BC (2002) Caspofungin in combination with itraconazole for the treatment of invasive aspergillosis in humans. Clin Infect Dis 34:1160–1161
Aliff TB, Maslak PG, Jurcic JG, Heaney ML, Cathcart KN, Sepkowitz KA, Weiss MA (2003) Refractory Aspergillus pneumonia in patients with acute leukemia: successful therapy with combination caspofungin and liposomal amphotericin. Cancer 97:1025–1032
Wheat LJ (2003) Combination therapy for aspergillosis: is it needed, and which combination? J Infect Dis 187:1831–1833
Groll AH, De Lucca AJ, Walsh TJ (1998) Emerging targets for the development of novel antifungal therapeutics. Trends Microbiol 6:117–124
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Groll, A.H., Kolve, H. Antifungal Agents: In Vitro Susceptibility Testing, Pharmacodynamics, and Prospects for Combination Therapy. Eur J Clin Microbiol Infect Dis 23, 256–270 (2004). https://doi.org/10.1007/s10096-004-1108-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-004-1108-6