
Abstract
The objective was to evaluate time to reach an EDSS of 4, 6, and 7 in NMOSD and MOGAD patients included in the Argentinean MS and NMOSD registry (RelevarEM, NCT 03,375,177).
Methods
NMOSD patients diagnosed according to 2015 criteria and with MOGAD were identified. Patients with at least 3 years of follow-up and periodic clinical evaluations with EDSS outcomes were included. AQP4-antibody and MOG-antibody status was recorded, and patients were stratified as seropositive and seronegative for AQP4-antibody. EDSS of 4, 6, and 7 were defined as dependent variables. Log rank test was used to identify differences between groups.
Results
Registry data was provided for a total of 137 patients. Of these, seventy-five presented AQP4-ab-positive NMOSD, 45 AQP4-ab-negative NMOSD, and 11 MOGAD. AQP4-ab status was determined by cell-based assay (CBA) in 72% of NMOSD patients. MOG-ab status was tested by CBA in all cases. Mean time to EDSS of 4 was 53.6 ± 24.5 vs. 63.1 ± 32.2 vs. 44.7 ± 32 months in seropositive, seronegative NMOSD, and MOGAD, respectively (p = 0.76). Mean time to EDSS of 6 was 79.2 ± 44.3 vs. 75.7 ± 48.6 vs. 54.7 ± 50 months in seropositive, seronegative NMOSD, and MOGAD (p = 0.23), while mean time to EDSS of 7 was 86.8 ± 54 vs. 80.4 ± 51 vs. 58.5 ± 47 months in seropositive, seronegative NMOSD, and MOGAD (p = 0.39).
Conclusion
No differences were observed between NMOSD (seropositive and seronegative) and MOGAD in survival curves.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuromyelitis optica spectrum disorders (NMOSD) are rare but often devastating inflammatory diseases of the central nervous system (CNS) characterized by severe attacks of transverse myelitis (TM), optic neuritis (ON) and/or brainstem syndrome [1, 2]. Myelin oligodendrocyte glycoprotein antibody (MOG-ab)-associated disease (MOGAD) often present with similar NMOSD attacks at disease onset, particularly in adult patients [3]. Disease presentation follows a relapsing course in up to 90% of NMOSD patients and in up to 80% of adult MOGAD patients, while progressive forms are uncommon in both conditions [1, 4, 5]. In NMOSD and MOGAD, disability typically accumulates with each clinical attack, resulting in long-term impairment of motor and/or visual function, as well as affecting other organ systems [1]. Currently, MOGAD is considered a separate nosologic entity pathogenetically distinct from both aquaporin4-antibodies (AQP4-ab)-positive NMOSD and multiple sclerosis (MS) [6,7,8]. Short- and long-term prognosis in patients suffering NMOSD or MOGAD is still uncertain [1, 6,7,8,9].
Recently, we presented the methodology behind RelevarEM, the first nationwide MS registry in Argentina and Latin America (NCT03375177) [10, 11]. The registry collects information about MS and NMOSD patients regarding sociodemographics, comorbidities, disability as measured by the Expanded Disability Status Scale (EDSS) score, relapse frequency, acute and long-term treatments, imaging (MRI) and cerebrospinal fluid (CSF) findings [10, 11].
The objective of this study was to evaluate time to EDSS scores of 4, 6, and 7 in NMOSD and MOGAD patients included in the Argentinean MS and NMOSD registry.
Methods
A retrospective study was conducted in a cohort of NMOSD and MOGAD patients followed in specialized MS/NMOSD centers from Argentina and enrolled in RelevarEM, a nationwide, longitudinal, observational, non-mandatory registry of MS and NMOSD patients (https://www.latambase.com.ar/login). Details related to RelevarEM procedures and methods have been previously published elsewhere [10, 11]. One of the goals of the registry is to create a network of neurologists involved in caring for MS/NMOSD patients in Argentina and to collect standardized relevant information from them on: routine clinical practice (at baseline: disease onset, course, symptoms, recovery, serological test, and methodology used); patient demographics (at baseline: patient identification, center, informed consent, administrative information); clinical findings (date, EDSS, bouts, paraclinical tests at follow-up: MRI date, MRI new lesions, CSF findings); immunotherapy prescribed (treatment used for NMOSD/MOGAD and safety: adverse events); and outcomes observed [10, 11]. To reduce risk of selection bias, neurologists participating in this registry were required to register all patients followed in their clinical practice.
For the study, registry neurologists treating patients with phenotypes suggestive of NMOSD and MOGAD were invited to send information on any patient with confirmed NMOSD diagnosis according to 2015 NMOSD criteria [1], or with MOGAD, with a diagnosis based on core clinical characteristics and presence of serum MOG-ab [6, 7]. Only patients with at least 3 years of follow-up and periodic clinical evaluations (at least two per year) with EDSS outcomes were included in the analysis. Attacks were assessed retrospectively. To ensure homogeneous data collection, we designed a specific web-based platform to investigate NMOSD and MOGAD attacks and EDSS.
To further classify patients, AQP4-ab and MOG-ab statuses were also recorded at any time over the disease course. AQP4-ab status was determined by cell-based assay 2 (CBA) and tissue-based indirect immunofluorescence (Table 1) [12, 13]. MOG-ab levels were tested using CBA in all cases [13] (Table 1).
Statistical analysis
Quantitative patient variables were analyzed descriptively. The Kruskal–Wallis test (non-parametric test) was used to estimate differences among the three groups. EDSS score of 4, 6, and 7 were defined as dependent variables. Time to events were estimated using Kaplan–Meier survival analyses. Log rank test was used to identify differences between groups (NMOSD seropositive, seronegative, and MOGAD). Multivariable Cox regression models were created to assess the independent predictive value of demographic and clinical characteristics to EDSS milestones. P < 0.05 was considered significant. We used STATA (Data Analysis and Statistical Software; StataCorp, College Station, TX) for the analyses.
Results
Registry data was provided on a total of 137 patients of which 131 had data about AQP4-ab or MOG-ab status (Table 1). Of these, seventy-five presented positivity for AQP4-ab; forty-five were seronegative; and 11 MOGAD. AQP4-ab status was determined by CBA in 72% of NMOSD patients, followed by IIF in 27%. Median age at disease onset was 38 ± 5, 41 ± 6.5 and 36 ± 8 years for each of the three groups, and disease duration was 6.1 (± 3), 6.0 (± 3.1) and 3.1 (± 2.1) years for seropositive, seronegative NMOSD, and MOGAD, respectively (Table 1).
The most frequent first treatment used to control the disease was azathioprine in NMOSD (seropositive and negative) as well as in MOGAD at disease onset, while rituximab was the most currently used treatment (Table 2). Only 1 (1.3%) patient in seropositive NMOSD and 2 (4.4%) in seronegative NMOSD never received treatment to control the disease.
Survival curves
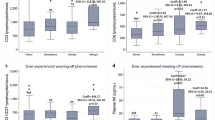
Mean time to EDSS of 4 was 53.6 ± 24.5 vs. 63.1 ± 32.2 vs. 44.7 ± 32 months in seropositive, seronegative NMOSD, and MOGAD, respectively (p = 0.76). Mean time to EDSS of 6 was 79.2 ± 44.3 vs. 75.7 ± 48.6 vs. 54.7 ± 50 months in seropositive, seronegative NMOSD, and MOGAD (p = 0.23), while mean time to EDSS of 7 was 86.8 ± 54 vs. 80.4 ± 51 vs. 58.5 ± 47 months in seropositive, seronegative NMOSD, and MOGAD (p = 0.39) (Fig. 1).

Survival curves to EDSS milestones in patients included
Cox regression analysis showed that the most relevant clinical factor associated with higher risk of EDSS was the presence of relapses during follow-up (HR 4.8 95% CI 2.2–7.9, p < 0.001), followed by age at disease onset (HR 2.15, 95% CI 1.22–2.88, p = 0.002) and higher EDSS after the first relapse (HR 1.41, 95% CI 1.17–2.15, p = 0.004). No association was observed with gender and diagnosis, and a trend in the reduction of the risk was observed with less time from disease onset to treatment initiation (Table 3). Due to the retrospective and exploratory nature of the study, no adjustment for multiple comparisons was performed.
Discussion
This is the first study performed in Latin America to evaluate disability outcomes in NMOSD and MOGAD. Mean time to EDSS of 6 was 76 months in NMOSD and 54 months in MOGAD. MOGAD patients accumulated the disability mostly in the first year of follow-up. Relapses were the most relevant factor related with higher risk of disability outcomes, and no differences were observed between NMOSD (seropositive and seronegative) and MOGAD in survival curves.
Unlike MS, neurologic disability is typically relapse-dependent and accumulates with each clinical attack [1]. Thus, 50% of NMOSD patients will require the use of a wheelchair or become functionally blind within 5 years of the first attack [14]. Additionally, attacks in NMOSD patients also reduce life expectancy [15] when primary attacks involve the brainstem or when cervical lesions extend to the medulla; therefore, NMOSD attacks require prompt evaluation and timely treatment to restore function and mitigate disability [1, 9]. On the other hand, 60% of adult MOGAD patients will develop permanent neurological disability, including motor and visual deficits [16]. Recently, we evaluated 262 attacks and 270 therapeutic interventions in 131 NMOSD and MOGAD patients followed and treated in Argentina [17]. Regarding relapses and disability accumulation, in this cohort, we observed that complete remission of relapses were higher after the first attack when compared to overall attacks (lower complete remission rates in subsequent attacks). In addition, we also observed that age at disease onset was an independent predictor of NMOSD sequelae (seropositive and seronegative, as well as in MOGAD) [17].
Another factor in the present study related to higher risk of disability was the older age at disease onset. As previously demonstrated, late onset-NMOSD patients experienced significantly worsened disability over a short time [18,19,20,21]. This worsened prognosis among these patients is not exclusively associated with positivity for AQP4-ab, which is consistent with other reports from Europe and Asia [18,19,20,21,22]. A multicenter Korean study reported that AQP4-ab-positive late onset-NMOSD patients experienced a lower rate of relapse, subsequent risk of myelitis attacks, and a trend towards higher risk of severe disability (EDSS 6.0) [23]. In this LATAM cohort, late onset-AQP4-ab-positive NMOSD patients had higher EDSS scores than early onset NMOSD patients [5.1 (± 2.1) vs. 2.1 (± 1.3)], and age at onset was significantly correlated with worse disability, as measured through the EDSS score [18]. A possible explanation for the worse prognosis among late onset NMOSD patients may be that most of the increased frequency of myelitis in this subgroup (LETM on spinal cord MRI) at presentation [18,19,20,21,22]. This is likely to be related to the low rate of improvement over time or even to the possibility of death that increased the risk to reach disability milestones sooner [18,19,20,21,22].
Finally, another factor related to an increased risk of disability in our study was the presence of higher EDSS since the first relapses.
In our study, no differences in survival curves were observed between seropositive and negative NMOSD patients.
All these aforementioned factors should be taken into consideration at disease onset in NMOSD and MOGAD patients to individualize treatment and medical care with the aim of avoiding disease activity and the increased risk of disability during follow-up.
Our study has several limitations, including the retrospective design, the absence of an external evaluation to confirm a relapse and the low number of included patients in the MOGAD arm. It is also important to mention that patients were followed for a few years and a difference at last follow-up time was observed between NMOSD and MOGAD that could bias results. Nonetheless, the standardized collection of the data, the specialists involved in data collection, and the power of the study in terms of the number of included subjects in the NMOSD arm strengthen the observation. Another limitation is the EDSS scale used to measure disability. EDSS, which was originally developed for use in MS, is heavily focused on ambulation disturbances and does not sufficiently reflect visual deficits (e.g., NMOSD patients with isolated ON cannot achieve an EDSS score higher than 4, even if complete bilateral visual loss is present) [24].
In conclusion, in this cohort, the mean time to reach an EDSS of 4, 6, and 7 was 50, 70, and 80 months, respectively. No differences were observed between NMOSD (seropositive and seronegative) and MOGAD in survival curves. To the best of our knowledge, this is the first large cohort study from a Latin American region. Therefore, our study provides relevant data for clinical practice.
References
Wingerchuk DM, Banwell B, Bennett JL et al (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85(2):177–189
Waters PJ, McKeon A, Leite MI, et al. (2012) Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 78(9): 665–71; discussion 9.
Marignier R, Hacohen Y, Cobo-Calvo A et al (2021) Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol 20(9):762–772
Cobo-Calvo A, Ruiz A, Maillart E et al (2018) Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology 90(21):e1858–e1869
Carnero Contentti E, Rojas JI, Cristiano E, et al. (2021) Latin American consensus recommendations for management and treatment of neuromyelitis optica spectrum disorders in clinical practice[Mult Scler Relat Disord. 2020 Oct;45:102428]. Mult Scler Relat Disord 52:103026
Jarius S, Paul F, Aktas O et al (2018) MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. Nervenarzt 89(12):1388–1399
Jurynczyk M, Jacob A, Fujihara K, Palace J (2019) Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations. Pract Neurol 19(3):187–195
Sato DK, Callegaro D, Lana-Peixoto MA et al (2014) Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 82(6):474–481
Kleiter I, Gahlen A, Borisow N et al (2016) Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol 79(2):206–216
Rojas JI, Alonso Serena M, Garcea O et al (2020) Multiple sclerosis and neuromyelitis optica spectrum disorders in Argentina: comparing baseline data from the Argentinean MS Registry (RelevarEM). Neurol Sci 41(6):1513–1519
Rojas JI, Carra A, Correale J et al (2019) The Argentinean multiple sclerosis registry (RelevarEM): methodological aspects and directions. Mult Scler Relat Disord 32:133–137
Lennon VA, Wingerchuk DM, Kryzer TJ et al (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364(9451):2106–2112
Waters P, Woodhall M, O’Connor KC et al (2015) MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm 2(3):e89
Kitley J, Leite MI, Nakashima I et al (2012) Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 135(Pt 6):1834–1849
Mealy MA, Kessler RA, Rimler Z et al (2018) Mortality in neuromyelitis optica is strongly associated with African ancestry. Neurol Neuroimmunol Neuroinflamm 5(4):e468
Jarius S, Ruprecht K, Kleiter I, et al. (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation 13(1): 279
Contentti EC, Lopez PA, Pettinicchi JP et al (2021) Assessing attacks and treatment response rates among adult patients with NMOSD and MOGAD: Data from a nationwide registry in Argentina. Mult Scler J Exp Transl Clin 7(3):20552173211032336
Carnero Contentti E, Daccach Marques V, Soto de Castillo I, et al. (2020) Clinical features and prognosis of late-onset neuromyelitis optica spectrum disorders in a Latin American cohort. J Neurol 267(5): 1260–8
Zhang LJ, Yang LN, Li T et al (2017) Distinctive characteristics of early-onset and late-onset neuromyelitis optica spectrum disorders. Int J Neurosci 127(4):334–338
Mao Z, Lu Z, Hu X (2014) Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 83(12):1122
Cai LJ, Zhang Q, Zhang Y et al (2020) Clinical characteristics of very late-onset neuromyelitis optica spectrum disorder. Mult Scler Relat Disord 46:102515
Carnero Contentti E, Daccach Marques V, Soto de Castillo I, Tkachuk V, Lopez PA, Rojas JI (2020) Age at onset correlate with disability in Latin American aquaporin-4-IgG-positive NMOSD patients. Mult Scler Relat Disord 44: 102258.
Seok JM, Cho HJ, Ahn SW et al (2017) Clinical characteristics of late-onset neuromyelitis optica spectrum disorder: a multicenter retrospective study in Korea. Mult Scler 23(13):1748–1756
Jarius S, Paul F, Weinshenker BG, Levy M, Kim HJ, Wildemann B (2020) Neuromyelitis optica. Nat Rev Dis Primers 6(1):85
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
Ethics committee approval was obtained for each participating center, and a written informed consent (according to each committee, if necessary) was obtained from all participants before data collection.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rojas, J.I., Pappolla, A., Patrucco, L. et al. Disability outcomes in NMOSD and MOGAD patients: data from a nationwide registry in Argentina. Neurol Sci 44, 281–286 (2023). https://doi.org/10.1007/s10072-022-06409-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-022-06409-w