
Abstract
Peripheral neuropathy (PN) is a common neurological problem defined as a dysfunction of sensory, motor, and autonomic nerves. The presence of peripheral neuropathy has recently been noticed in Parkinson’s disease (PD) This comorbidity is concerning as it increases the burden on patients whose motor functions are previously compromised. A comprehensive computer-based literature review utilizing multiple peer-reviewed databases (e.g., Embase, PsycINFO, CINAHL, etc.) was conducted. There is evidence for the utility of robust diagnostic criteria to distinguish between large fiber neuropathy (LFN) and small fiber neuropathy (SFN). Some studies have established links between prolonged l-DOPA exposure and prevalence with increased levels of homocysteine (HCY) and methylmalonic acid (MMA) as pathological underlying mechanisms. PN in PD patients with relatively truncated exposure to l-DOPA therapy may have underlying mutations in the Parkin and MHTFR gene or separate mitochondrial disorders. Vitamin B12 and cobalamin deficiencies have also been implicated as drivers of PN. Accumulation of phosphorylated α-synuclein is another central feature in PN and deems urgent exploration via large cohort studies. Importantly, these underlying mechanisms have been linked to peripheral denervation. This review delves into the potential treatments for PN targeting B12 deficiencies and the use of COMT inhibitors along with other novel approaches. Avenues of research with powerful randomized controlled and long-term cohort studies exploring genetic mechanisms and novel treatment pathways is urgently required to alleviate the burden of disease exerted by PN on PD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Peripheral neuropathy (PN) is a common neurological problem defined as a dysfunction of peripheral motor, sensory, and autonomic nerves [1,2,3]. PN manifests as a disease of axons and/or myelin within nerve fibers [4]. Symptoms of PN may be caused by sensory disturbances preceding motor disturbances. However, other neuropathies such as chronic inflammatory demyelinating neuropathy may have motor disturbances as the predominant symptoms [5]. PN has many different etiologies including metabolic and endocrine disorders, genetic diseases, HIV, infectious diseases, and thyroid disease. The most common cause of PN is diabetes mellitus, with 60% of diabetes patients suffering from PN. [3]. Idiopathic peripheral neuropathy (IPN) has a very slow progression rate and is often seen in the older population [6].Other causes currently under review include genetic mutations in certain genes, i.e., Parkin gene for Parkinson’s disease (PD), deficiency of vitamin B12, folate, and vitamin B6 and B1 [6,7,8].
Idiopathic Parkinson’s disease (IPD) is a multisystem disease involving a progressive loss of dopaminergic innervation in various regions of the brain. IPD manifests itself with motor symptoms such as rigidity, gait, and balance problems, bradykinesia, and tremors as well as non-motor symptoms such as cognitive, autonomic, and sensory impairments [7]. The presence of sensory disturbances during the “off” medication state in PD, which has only recently been observed, points to a possible pathological involvement of PN in the disease [7]. However, much of the interest in peripheral neuropathy in PD has stemmed from observations in patients treated with duodopa or levodopa-carbidopa intestinal gel (LGIC). The presence of neuropathy in PD patients is a call for concern, as it would increase the burden on patients whose motor functions are already severely compromised [7].
Current scientific literature has brought up new discussions regarding the increased prevalence of PN in relation to PD. New literature suggests PN can cause balance problems and muscle weakness; thus, its presence can negatively affect the lives of PD patients [1, 9, 10]. Casting a wide net and examining the literature, it is clear that there are gaps with respect to reviews coalescing all the evidence of PN in PD patients. One major systematic review pools the relevant findings of PN and its impact and relationship to PD, which provides the foundations for the current qualitative review [1]. However, this review is quite novel since it is qualitative in nature and explores other important aspects of PN in PD, which have yet to be investigated. Within, the literature, it is currently unknown whether they are any effective treatments for PN in PD patients. We delve into this important yet seldom discussed aspect of treatment and explore the various animal models and current therapeutic measures underlying the attenuation of non-PD neuropathies and its potential for forging novel treatments in PN within the context of PD. As a result, these novel discussions should set our review apart from similar papers in the field. There is considerable evidence within the field of movement disorders regarding the relationship between large fiber neuropathy (LFN) and PD [1]. However, this current review dives into the seldom discussed small fiber neuropathy (SFN) and its etiologies, proposed pathological mechanisms supplemented by comparisons to LFN and insights into its impact on PD patients. Moreover, this qualitative review will discuss the etiology of peripheral neuropathy and the current treatment options present for PD patients suffering from such comorbidity.
Methods
A thorough and comprehensive search for English and non-English peer–reviewed articles indexed in MEDLINE, CINAHL, CENTRAL, Embase, and PsycINFO was made using multiple combinations of the following keywords: “peripheral neuropathy,” “polyneuropathy,” “peripheral denervation”, “Parkinson’s disease,” “PD,” “PN,” “diagnosis,” “treatment,” and “mechanism.” Based on a selection criteria tailored towards the study, publications from inception of the respective databases to 2020 were selected after careful analysis and a meticulous risk of bias assessment. Articles that dealt with PN in diseases other than PD were excluded from the literature search.
Results
Diagnosis of peripheral neuropathy
Unlike PD which can be diagnosed via clinical criteria, affirming a diagnosis of PN may require additional reinforcements such as the testing of peripheral nerves using nerve conduction studies and electromyography [1, 11, 12]. Even then, some neuropathies such as small fiber neuropathies (SFN) present difficulty in detection, often falsely presenting with normal results on nerve conduction studies. As such, robust clinical criteria become a necessity. Patients must display (i) 2 or more of the symptoms associated with SFN (pain, paresthesias, allodynia, thermal sensory loss, pinprick loss, sheet or sock intolerance, restless leg syndrome, sicca syndrome, accommodation problems, hyper/hypohydrosis, micturition disturbances, impotence and/or diminished ejaculation or lubrication, bowel disturbances, hot flushes, orthostatic dizziness, and cardiac palpitations), as well as (ii) a reduced intraepidermal nerve fiber density (IENFD) in skin biopsy or a pathological thermal threshold in quantitative sensory testing (QST) in order to get a correct diagnosis of SFN [12]. A thorough and complete patient history may provide the most useful information pertaining to a diagnosis by giving insight to strong contributing factors (i.e., alcohol use, heredity, new medications, and especially diabetes) [11]. The American Academy of Neurology (AAN) has diagnostic guidelines that support the use of a fasting glucose/2-h glucose tolerance testing, vitamin B12 testing with methylmalonic acid/homocysteine levels, and serum protein electrophoresis with immunofixation electrophoresis testing for initial investigation of peripheral neuropathy [11]. In cases where atypical symptoms are present, additional tests may be administered.
If SFN is suspected, intraepidermal nerve fiber density or analysis of quantitative sensory testing can help with diagnosis along with nociceptive evoked potentials. Skin biopsy is often a good, minimally invasive and inexpensive investigative tool to quantify epidermal nerve endings, distal ends of axons and trigeminal ganglia that cross the dermoepidermal junction and terminate within the epidermis [1, 12]. Autonomic testing utilizing a variety of reflex tests, intraepidermal electrical stimulation (IES) tests, corneal confocal microscopy, and microneurography may also contribute to the detection of changes in peripheral fibers in SFN and assist in a more affirmative diagnosis [12]. Recently, the noninvasive meta-iodobenzylguanidine (MIBG) myocardial scintigraphy has become widely popular in differentiating and detecting PD from other movement disorders. Cardiac sympathetic nerve fibers are typically found to be affected early on in the course of PD and this PN is picked up by the highly sensitive MIBG imaging tool [13].There is no official standard for determining the severity of SFN, but the use of aforementioned tools and precise questionnaires are useful to determine the span and severity of SFN-related sensory and autonomic symptoms [12].
Common Parkinson’s disease therapies and peripheral neuropathy
Levodopa
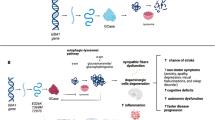
Levodopa (l-DOPA) is considered the cornerstone of therapy for PD [6]. Long-term l-DOPA therapy has shown to be effective in most patients. However, it causes significant motor fluctuations, including dyskinesia and early morning dystonia [14]. PN in PD patients has been attributed to extended l-DOPA use as it results in increased levels of homocysteine (Hcy) and methylmalonic acid (MMA). Fundamentally, this spike in Hcy and MMA results from the metabolism of l-DOPA in the O-methylation pathway [8]. Catechol-O-methyltransferase (COMT) converts l-DOPA into 3-O-methyldopa; this conversion requires a methyl-group which is donated by S-adenosylmethionine (SAM). Once the methyl group is donated, SAM converts into S-adenosylhomocysteine (SAH) and then, almost immediately into homocysteine [7, 8]. In normal circumstances, homocysteine is remethylated into methionine and SAM by methyl-THF and cobalamin or converted into cysteine via irreversible transsulfuration [8]. However, the methylation of l-DOPA results in the depletion of SAM, leading to increased levels of plasma homocysteine [7]. Vitamin B12 and l-DOPA have an inverse relationship for reasons that are not currently clear. This deficiency in cobalamin is thought to result in increased levels of Hcy and MMA as vitamin B12 is an essential cofactor for the conversion of homocysteine to methionine and SAM. Cobalamin also serves as a cofactor for the isomerisation of succinyl-CoA, and thus cobalamin deficiency also leads to MMA accumulation [7, 8]. Homocysteine elevation as a marker for reduced methylation capacity also has additional consequences other than polyneuropathy. Markedly and mildly elevated concentrations of circulating Hcy are associated with increased risks of vascular occlusion [15]. Elevated level of the nonprotein amino acid Hcy has been reported in the literature to be a risk factor for cardiovascular diseases, neurodegenerative diseases, and neural tube defects [16,17,18].
Several large studies have shown elevated Hcy and MMA levels in PD patients compared to controls, with greater elevation in patients with longer l-DOPA usage. In a multicenter study with 330 PD patients, the risk of neuropathy in PD patients with l-DOPA exposure was tested. 19.4% of PD patients with long exposure (> 3 years) to l-DOPA showed neuropathy while only 6.8% of patients with short exposure (< 3 years) showed neuropathy; 4.82% of PD patients without l-DOPA exposure were found to have neuropathy [12]. Healthy controls with similar age were diagnosed with axonal neuropathy, predominantly sensory at 8.76%. Among the patients with long term l-DOPA usage that were diagnosed with neuropathy, vitamin B12 levels were generally lower while Hcy and MMA levels were significantly elevated [12]. Risk of neuropathy, in this study, was also noted to increase by 8% each year of age, indicating the risk factors of PN in PD patients are not limited to duration of l-DOPA use [12]. In contrast, other studies, with age controlled trials, have found age is not a variable for neuropathy in PD; however, age-related vitamin B12 plays a role along with other age-related nutritional deficiencies [6].
Another similar study involving l-DOPA therapy and the prevalence of PN in PD patients found only 12.1% of their patient population that was l-DOPA naïve had PN, compared with 36.1% of l-DOPA-treated PD patients with PN [6]. The significant increase in prevalence suggests l-DOPA therapy plays a crucial role in the development of PN in PD patients. Yet again, the results indicated decreased vitamin B12 levels along with decreased folate levels. Reduced folate levels (< 10 mg/L) independently raise the risk of neuropathy three times above the normal lower limit. Independent association of neuropathy accompanied by low folate levels have also been connected to other disorders such as cognitive impairment and depression, both of which are common in PD patients [6]. Alternatively, these findings may not indicate that low folate levels are directly responsible for peripheral nerve damage but rather that low folate levels may indicate other nutritional deficiencies that may play a role in neuropathy.
Conversely, research conducted by Reynolds et al. revealed clinical and experimental evidence supporting the finding that in the presence of vitamin B12 deficiency, prolonged excess folate can result in neurological deterioration, which is implicated in PD [19]. It seems that the risk of neurological relapse may be greater as the dose of excess folic acid is increased. Additionally, a study was carried out by Brito et al. [20] in an effort to better characterize the human serum metabolome in vitamin B12 deficiency and its affiliation with neurological function. The results revealed that vitamin B12 status is associated with serum metabolic markers of myelin integrity in the central nervous system, mitochondrial function, oxidative stress, and peripheral nerve function. By extension and in relation to other studies, these findings imply that a vitamin B12 deficiency may be correlated with overall decreased neurological function, and as such, be implicated in PD. Further studies are required in order to determine whether vitamin B12 and folate deficiencies cause neuropathy or whether they are simply one element in a host of elements contributing to PN [6]. Furthermore, additional research should pioneer the investigation of the utility of substituting other B vitamins (ex. B1, B6, etc.) besides vitamin B12 and folate.
Many recent studies have implicated cobalamin deficiency due to L-DOPA therapy as a potential cause for neuropathy, while others implicate folate deficiency. In one study, a cohort of l-DOPA-treated PD patients was administered monthly intramuscular injections of vitamin B6 and vitamin B12 [21]. This cohort had higher 3-OMD levels than cohorts that were treated with either l-DOPA with dopamine decarboxylase inhibitor or a duodopa regimen. However, no significant differences were found for Hcy levels, oral LD dosage or LD concentrations [21]. The study does not answer the question of whether methyl-donating vitamins increase l-DOPA metabolism, nor was PN considered. An alternative hypothesis proposed by Toth et al. suggests that PN may not be directly caused by vitamin B12 deficiency, but may be related to cobalamin instead due to the increase in neurotoxic metabolites such as MMA and Hcy [7]. In their study, they observed that all PN symptoms arose well after the onset of PD and initiation of l-DOPA therapy. The Toronto Clinical Scoring System (TCSS) measured a positive correlation between PD patients with IPN (PD-IPN) and L-DOPA usage time suggesting that PN is treatment related [7]. In order to determine the role of MMA and Hcy along with cobalamin in PD, PD-IPN, and PD patients with determined cause of peripheral neuropathy (PD-DPN) were tested for MMA and Hcy levels. A number of patients with PD-IPN and IPN concomitant cobalamin deficiency (IPN/CD) had cobalamin levels higher than 300 pg/mL yet had abnormal levels of Hcy and MMA [7]. However, PD-IPN and PD-DPN had similar levels of cobalamin indicating that cobalamin levels do not explain PN in PD-IPN patients. Furthermore, PD only patients with less lifetime exposure to l-DOPA also had normal cobalamin levels, indicating that abnormal levels of neurotoxin metabolites such as Hcy and MMA may be related to the development of PN instead of vitamin B12 deficiency [7, 22].
A feature that is common in all of the aforementioned studies is that only a percentage of PD patients that undergo short or long-term l-DOPA therapy exhibit neuropathy. This may be associated with genetic susceptibility for the development of neuropathy in some patients. For instance, one-third of the patients identified with Hcy elevation had impaired methylenetetrahydrofolate reductase enzyme, which is responsible for the remethylation of homocysteine in methionine [7].
Several studies have reported the emergence of PN in PD patients from a continuous duodopa regimen. Herein, we consider PN in l-DOPA and duodopa patients separately given the evidence for clinical differences between these groups. Duodopa therapy or levodopa-carbidopa intestinal gel (LCIG) is a PD therapeutic involving the dual fusion of carbidopa and l-DOPA delivered through the enteric nervous system. Duodopa may be used to control for more severe motor complications when oral therapy is ineffective. Patients treated with duodopa tend to have higher daily doses of l-DOPA, longer disease duration, and higher incidence of peripheral neuropathy [13, 23, 24]. In a study comparing PN and B vitamin levels in 13 patients treated with l-DOPA and 8 treated with duodopa, who were matched for age, Hoehn and Yahr stage, and UPRDS III, all LCIG patients developed PN (mostly axonal sensory or sensorimotor), whereas only 8 out of 11 l-DOPA-treated patients developed PN [25]. PN in orally treated patients was also milder, with fewer damaged nerves. There were no significant differences between groups on B-vitamin levels, however [25].
In a case study by Urban et al., a 67-year-old male diagnosed with PD for 5 years, and a 76-year-old female diagnosed with PD for 23 years underwent a sole duodopa regimen [26]. After 13 months of duodopa treatment, both developed axonal sensorimotor PN, and cobalamin and vitamin B6 deficiency, which were later stabilized by parenteral substitution treatment. This suggests, as the authors concluded, that PN emergence might have been due to lack of cobalamin and vitamin B6 [26]. Meppelink et al. found that 2 of their 15 patients on duodopa developed axonal PN. PN symptoms were attenuated when the duodopa dose was lowered, and vitamin B12 supplements were administered [27]. In addition, there have been several anecdotal reports of PD patients exhibiting PN which clinically resembles Guillain-Barré syndrome (GBS) from continuous duodopa therapies [28,29,30].
One prospective study by Merola et al. involved a duodopa treatment regimen in 15 PD patients with an average follow-up of 9 months. Upon a follow-up, 9 of their participants expressed a mild axonal length-dependent PN akin to that arising from duodopa therapy. Three of the 9 participants already had PN at baseline relating to a previous l-DOPA regimen, though the symptoms of PN worsened with duodopa [31]. In an extension of the same protocol, 33 PD patients were given LCIG therapy and were followed for 24 months. Two patients developed subacute PN, 2 developed chronic PN, and 7 developed subclinical PN, with chronic PN patients having a higher l-DOPA equivalent daily dose. When these patients were given vitamin B1 and B12 supplements, all showed clinical improvement [23]. A large international, 12-month open-label study on duodopa therapy in 354 PD patients found 6% of them presented with PN, of which 0.6% had characteristics similar to GBS [32]. Furthermore, a study on 5 PD cases being treated with duodopa therapy developed axonal PN, folate deficiency, and vitamin B12 deficiency, suggesting a possible link between the deficiencies of these methyl group donors with the duodopa regimen [33]. More recently, a study by Rispoli et al. with 30 LCIG patients supplemented with vitamin B reported a 19% prevalence of emergent PN. Four of the 21 patients, who had normal electrophysiological assessment at baseline, developed symmetrical axonal PN [34]. All patients in the study displayed worsening conduction in the sural sensory and peroneal motor nerves at a long-term follow-up of 42.4 months [34]. However, the lack of a control group limits the ability of the study to draw conclusions about the effect of B vitamin substitution on PN in PD.
Neuropathy of small fibers has also been observed in patients treated with LCIG, although the number of studies investigating this comorbidity is limited. One study, which compared 6 l-DOPA naïve patients, 6 oral l-DOPA patients and 5 advanced PD patients starting l-DOPA found that all patients starting l-DOPA had significant damage to small nerve fibers and an increased thermal threshold [35]. After 3 months, axonal swelling was observed, and at 6 months, almost complete epidermal and dermal denervation occurred [35]. These results demonstrate a need for monitoring of small fiber neuropathy in LCIG.
Taken together, these findings demonstrate a strong relationship between polyneuropathy and LCIG treatment in PD. However, there is much that is unresolved from the aforementioned studies demonstrating results of PN arising from duodopa therapy in PD. Is there a relationship between the PN arising from l-DOPA therapy, and the PN arising from duodopa therapy? Is the emergence of a PN characteristic of GBS in PD in anyway related to the typical PN that is seen from duodopa therapy in PD? Comparative studies examining these differences are needed. Given the heterogeneity in reported prevalence of PN in LCIG-treated patients, studies with larger sample sizes are also needed to reliably estimate the rate of comorbidity. Finally, while several studies have demonstrated a therapeutic role of B-vitamin substitution in managing PN in PD, the results are mixed. Currently, there is no systematic treatment yet for duodopa-induced PN in PD [36]. Hence, further studies are required in order to elucidate the therapeutic and associational profile of duodopa-induced PN in PD.
COMT inhibitors
Considering the pathophysiology of PN that is derived from the extended use of l-DOPA, a common conclusion for PN therapy would include the use of COMT inhibitors (COMT-Is). Hypothetically, COMT-Is should prevent the conversion of l-DOPA into 3-O-methyldopa to stop O-methylation pathway via the use of l-DOPA, thereby, effectively reducing Hcy and MMA levels. Lamberti et al. studied 78 patients in order to verify whether Hcy levels are increased in l-DOPA patients, and to assess the efficacy of COMT-Is on concentrations of Hcy [16]. The study included 26 patients that were treated with l-DOPA, 20 patients treated with l-DOPA and COMT-I, and 32 control patients. They discovered that patients with long-term l-DOPA use indeed showed increased Hcy levels. Furthermore, plasma Hcy levels were significantly higher in PD patients that endured l-DOPA therapy compared with those that endured l-DOPA therapy with COMT-I. Plasma levels had an inverse correlation with folate and cobalamin concentrations in all patients.
Furthermore, a 5-day study conducted by Nissinen et al. on animal models supported the findings mentioned above [24]. When healthy adult rats were treated with l-DOPA, results indicated a marked increase in l-DOPA-induced hyperhomocysteinemia. However, the administration of the COMT inhibitor, entacapone, significantly (p < 0.001) reduced the l-DOPA-induced rise in plasma Hcy levels for up to 2 h post-treatment on day 5 of the study. In yet another study, Zoccolella et al. evaluated the effects of different antiparkinsonian drugs on Hcy concentrations [37]. The study included 15 patients that were treated with dopamine agonists, 15 patients treated with l-DOPA, 15 patients treated with l-DOPA and COMT-I, and 15 control patients. The results indicated that the administration of l-DOPA significantly increased Hcy levels while the addition of COMT-I reduced Hcy concentrations.
In another study, however, COMT-I administration did not show a reduction in Hcy and MMA levels in IPD patients [22]. Although, COMT-Is show potential in reducing Hcy and MMA levels in some studies, larger studies are required in order to obtain confirmed results for the efficacy of COMT-Is.
Links between genetics and peripheral neuropathy?
Parkin gene
Genetic variants in the parkin gene have also been implicated with neuropathy in PD patients in single case reports. PD patients with sensory dominant axonal polyneuropathy have shown the presence of parkin gene fragments. However, a definite relationship between parkin mutations and PN remains unclear [7, 22, 38].
MTHFR gene
Another potential candidate gene involved in PN with PD may be the MTHFR gene. The MTHFR gene encodes its abbreviated namesake enzyme, methlenetetrahydrofolate reductase. This enzyme is involved in catalyzing methylenetetrahydrofolate to 5-methyltetrahydrofolate, which is a cosubstrate in the reaction pathway required to convert Hcy to methionine [39]. A study by Gorgone et al. found a TT677 mutation in the MTHFR gene is more frequently found in PD patients compared with controls. Furthermore, the TT677 MTHFR gene mutation was related to elevated Hcy levels in PD patients compared with controls. They also found a direct positive correlation between daily l-DOPA dose and Hcy levels, though it was independent of the previous correlation of the TT677 mutation with Hcy levels [39]. This suggests that the mutant MTHFR gene may not be directly involved with the l-DOPA-induced increases of Hcy that play a role in the risk of PN in PD. Further studies are required to investigate whether there is a potential indirect association between the MTHFR-TT677 mutation with PN in PD.
Mitochondrial disorders and peripheral neuropathy
Patients with PN and PD also present with co-occurrence of mitochondrial disorders [40]. The mitochondria are believed to interact with the PD marker of α-synuclein aggregations [40]. This interaction may result in impaired neuronal function to the basal ganglia, cerebellum, sympathetic ganglia, and the enteric nervous system [41,42,43]. Furthermore, many mitochondrial diseases show loss in peripheral nerves along with myopathy changes. Despite these known problems caused by mitochondrial disorders, there have not been any reports in the literature with respect to mitochondrial disorders and PN in PD patients [44, 45].
Denervation in Parkinson’s disease
Autonomic neuropathy is commonly found in patients suffering from PD. These non-motor displays of PD may cause the patient to suffer from orthostatic hypotension, delayed gastric emptying, impaired sweating, and sebum production [46, 47]. In some cases, a mild form of autonomic dysfunction may be seen within the early stages of PD, but autonomic denervation is usually considered to be a late complication of the disease thought to be complicated by PD medications [22, 48, 49]. Some studies however suggest otherwise.
A study undertaken by Dabby et al. assessed peripheral autonomic neural involvement in PD using a punch skin biopsy. The autonomic denervation of blood vessels, sweat glands, and arrector pili muscles was assessed. Comparing PD patients with controls the researchers found that denervation of the peripheral autonomic nervous system in the skin of PD patients was significantly higher than controls. Only 2 out of the 22 PD patients in this study returned results depicting a normal skin biopsy. The researchers also found no difference in skin biopsy scores between treated and untreated patient. Thus there may be no association between antiPD medications (particularly l-DOPA) and the denervation of autonomic structures [46]. Decreased cardiac uptake of MIBG has been reported in PD patients with Lewy bodies by an imaging approach using myocardial scintigraphy [50]. Cardiac sympathetic denervation is known to occur in Lewy body disease, although it is not specific for Lewy body disease. Furthermore, cardiac sympathetic denervation may account for decreased cardiac uptake of MIBG in PD [50, 51]. It is known that in the progression of PD early pathological change starts at the dorsal vagal nucleus, which is the lower part of the brainstem [52, 53]. When and how degeneration of the cardiac sympathetic nerve begins is still unclear. Orima et al. used immunohistochemistry to examine cardiac tissue, sympathetic ganglia, and medulla oblongata at dorsal vagal nuclei from patients who had incidental Lewy body disease (ILBD), which can be a presymptomatic stage of PD [54]. Anti-TH was used as a marker for sympathetic nerves. Orima et al. found that the TH-immunoreactive nerves had nearly disappeared in 6 out of 20 subjects examined. Immunoreactive nerve fibers of fascicles in the epicardium were preserved well in half of the subjects examined. Neuronal cell loss in the dorsal vagal nucleus and the sympathetic ganglia was not detected in any of the ILBD patients [54]. These findings suggest that degeneration of the cardiac sympathetic nerve starts in the early stages of PD and that it occurs before neuronal cell loss in the dorsal vagal nucleus.
Phosphorylated α-synuclein and peripheral neuropathy
Phosphorylated α-synuclein has been detected in the peripheral nervous system of patients with PD. Specifically, α-synuclein deposits have been found in the autonomic nerves of the colon, cardiac plexus, and more recently in the cutaneous C-fibers [47, 55,56,57]. Cumulative l-DOPA exposure has been proposed as a potential cause for large fiber neuropathy via Hcy accumulation and reduced levels of folate as well as vitamin B12 [6, 7, 12]. However, several studies have found no relation between peripheral denervation and l-DOPA exposure suggesting that peripheral nerve involvement may be an intrinsic feature of the disease, especially in relation to small fiber neuropathy [47, 49, 58]. Furthermore, the identification of α-synuclein in cutaneous nerves has led to the consideration of skin tissue as a potential biomarker in PD [55, 59]. Donadio et al. set out to understand whether α-synuclein deposits in the skin nerve fibers would be a useful biomarker of patients with IPD. Examining patients with IPD and patients with Parkinsonism assumed not to have α-synuclein deposits (PAR-vascular Parkinsonism, tauopathies), the researchers found that phosphorylated α-synuclein in proximal peripheral nerves is a sensitive biomarker for IPD diagnosis [55]. This is useful in differentiating IPD from other forms of Parkinsonism. Furthermore, neuritic inclusions of α-synuclein were correlated with a small-fiber dysfunction suggesting a direct role in peripheral nerve dysfunction. These neuritic inclusions of α-synuclein also differed in IPD and pure autonomic damage, suggesting different pathologies [55, 60]. Moreover, a crucial differentiating factor between IPD and PN was the site of analysis of the skin nerves [60]. In IPD, the location of deposit analysis is critical. Thus, the spatial distribution of α-synuclein deposits and the pattern of innervation may highlight the underlying differences in pathologies [60].
Pathogenic mechanisms of peripheral neuropathy
The exact pathogenic mechanism of PN is not clear as many factors may contribute to PN. In IPD, PN has been linked to the long-term use of l-DOPA, hence suggesting an iatrogenic connection. Long-term l-DOPA use has been linked to decreased cobalamin and increased Hcy and MMA levels. Elevated levels of plasma Hcy levels have been associated with increased vulnerability of mitochondrial toxins and rising free radicals, inducing inflammatory reactions and impaired DNA repair mechanisms [12, 59]. Hcy elevation has also been associated with sural axonal neurodegeneration in electrophysiological studies of patients with PD. Elevated levels of Hcy are also present in diabetic patients with PN and has been associated with peripheral nerve damage [12]. Elevated Hcy levels promote excitotoxicity via stimulation of N-methyl-D-Aspartate receptors and can damage neuronal DNA, resulting in apoptosis [61]. MMA elevation can result in lipid and protein oxidative damage in vitro. In vivo, MMA results in lipid peroxidation and inhibition of glutathione peroxidase [38, 61]. Furthermore, increased levels of MMA have been associated with abnormal odd chain and branched chain fatty acids with abnormal myelination, which can lead to defective nerve transmission [61].
Mitochondrial dysfunction and oxidative stress have been thought to contribute to the pathogenesis of PD. Animal models treated with mitochondrial inhibitors developed PD symptoms and pathology. Mitochondrial complex I deficits in PD patients are associated with increased free-radical production, which leads to apoptotic cell death along with increased vulnerability to the mitochondrial toxin 1-methyl-4-phenulpyridinium and the reactive metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [62, 63]. Although little research has been conducted on the pathogenesis of mitochondrial dysfunction in PD patients and its association with PN, there are many aspects that may be extrapolated from diabetic neuropathy. The pathophysiology of sensory neuropathy in diabetic patients may involve the activation of apoptosis associated with mitochondrial dysfunction. Apoptotic cell death as a result of mitochondrial dysfunction has also been seen in PD; hence, it may be implicated in neuropathy in PD patients [63, 64]. In both diabetic patients with neuropathy and patients with Parkinson’s disease, the apoptotic effector protein Bax has been implicated. Bax travels from the cytosol to the mitochondria during apoptosis and binds to the adenine nucleotide translocator and holds the pore open during apoptosis. Bax also destabilizes the outer mitochondrial membrane [15, 63,64,65].
The expression of parkin mRNA in human peripheral nerve may be responsible for axonal PN in individuals with parkin-related PD. Reported cases for parkin mutations include deletions in exon 3, exon 5, and missense mutations in exon 12. These cases were linked to early onset and prolonged disease duration, suggesting that PN develops in the later stages of PD [38].
The parkin gene encodes an E3 ubiquitin ligase that plays a role in proteasomal degradation. Exon 3 of parkin codes for a region of the parkin protein, which binds target proteins and proteasomes. Mutations in this region impair the recognition and ubiquitination of target proteins before proteasomal degradation and binding of the Rpn10 subunit of 26S proteasome. This loss of parkin function and proteasomal degradation impairment can result in the accumulation of neurotoxins in the peripheral nerve. Parkin mutation and its pathogenic role in the development of PN is not clear [38].
Treatments of peripheral neuropathy in Parkinson’s disease?
In one study, IPD patients with PN and cobalamin deficiency, MMA and Hcy elevated levels were given cobalamin injections of 1000 μg after the diagnosis of PN. Clinical examinations along with blood tests were performed for these patients at 6, 12, and 24 months to see whether cobalamin injections were having any effect. Within the first 6 months, all patients had normal levels of cobalamin while 2/3 of the patients presented with markedly improved levels of Hcy and MMA. These levels remained stable over the follow-up at 12 to 24 months along with clinical assessments and electrophysiological measurements. These observed results were only limited to PD patients with PN. When compared with PN patients without PD with only MMA elevation, mild clinical and electrophysiological decline was seen over 12 to 24 months with receiving cobalamin injections [1, 7]. There are limited studies concerning treatment of PN in PD. Larger studies are required to assess the efficacy of cobalamin injections along with folate supplements and COMT-Is, particularly in l-DOPA-treated patients [6].
Novel treatment approaches for PN within the setting of IPD is an area that requires urgent exploration [1]. Due to the comorbidity and burden of disease it exerts on patients, holistic and interdisciplinary care is required [1, 8, 66]. These types of holistic treatments can serve as important adjuncts to conventional treatment of PN. Case in point, electromagnetic and laser therapy is garnering evidence with respect to treatment of diabetic peripheral neuropathy [67]. Utilizing a visual analog scale and Toronto Clinical Neuropath Scoring System to assess pain and neuropathy, tailored treatments were implemented [67]. This particular RCT suggested (albeit nonsignificant findings) supplementing central therapy of diabetes with laser or magnetic therapy could prove to be beneficial [67]. As a result, RCTs extrapolating the efficacy of laser and magnetic therapy in alleviating PN and preventing further deterioration is warranted.
Currently, the mainstay of treatment for PN revolves around the use of COMT inhibitors such as entacapone with l-DOPA therapy [1, 68]. A multicenter study on PD patients with PN noted higher vitamin B12 levels (p < 0.0001) and lower serum HCY levels (p 1/4 0.001) [1, 68]. Interestingly, this cohort of patients who were administered COMT inhibitors were on l-DOPA therapy for at least 3 years [1, 68]. This is a powerful study that lends credence to the use of COMT inhibitors to alleviate l-DOPA-induced PN in IPD as evidenced by changes in relevant biomarkers (vitamin B12 and serum HCY levels). There are limitations that need to be addressed such as organic PN in IPD without l-DOPA exposure. Furthermore, COMT inhibitors would prove to be ineffective against other etiologies of PN such as due to genetic mutations, mitochondrial disorders and a-synuclein accumulation. Thus, novel approaches outside the realm of COMT inhibitors are required.
A systematic review examining the therapeutic efficacy of vitamin B12 and other complexes along with methylcobalamin on symptom relief and electrophysiological improvements was performed [69]. They noted combination of vitamin B12 and methylcobalamin administration alleviated pain and paresthesia but exerted inconsistent effects on electrophysiological assessments and vibration perception [69]. The authors promote a combination of B12 and methylcobalamin which may lead to greater symptomatic relief in diabetic neuropathy that modulations in electrophysiological results, but simultaneously call for powerful RCTs to further probe the underlying mechanisms of vitamin B12’s therapeutic efficacy [1, 69]. Once again, the caveat here is majority of RCTs and systematic reviews investigating management plans for PN center around diabetic neuropathy [1, 6, 66]. There is a dearth in the evidence with regard to trials examining therapeutic efficacy of various drugs on PN within the context of IPD [1].
Within the frontiers of experimental neurology, animal models probing the certain enzymes and genetic markers as therapeutic targets for PN have been promising. For example, inhibition of histone deacetylase 6 (HDAC6) in a mutant mouse model may attenuate Charcot-Marie-Tooth type 2A peripheral neuropathy [70]. This mouse model study may pave the way for future trials to develop drugs to build up on the inhibition of HDAC6 and extrapolate these therapeutic targets to IPD PN. Importantly, these genetic studies in animal models need to extend to the Parkin and MHTFR genes [1, 22, 39, 44]. Recently, there is a surge of studies investigating PN from an oncological perspective. This is due to PN stemming as an adverse effect from chemotherapeutic agents such as platinum [71, 72]. As a result, bendamustine-rituximab (BR) combination has demonstrated some therapeutic efficacy in alleviating immuno-mediated neuropathies in certain malignancies [1, 71, 72]. This indicates certain combinations of monoclonal antibodies that have worked well against other types of neuropathies may prove to be beneficial in IPD PN [1, 71, 72]. As PN is quite prevalent in diabetes, much of the research has emphasized treatments targeting diabetic neuropathy [1, 12, 73]. Case in point, another animal model study using diabetic rats noted cemtirestat (aldose reductase inhibitor) reduced PN through mechanisms entrenched in lowering triglycerides [73]. Other cellular and molecular studies have identified important voltage-gated sodium channels in peripheral nerves that are crucial therapeutic targets in diabetic neuropathy [73, 74]. When comparing ranolazine against pioglitazone, ranolazine performed better in targeting those aforementioned channels and exerting neuroprotective effects [74]. The next step would be to trial ranolazine and pioglitazone via RCTs in IPD patients suffering from PN as IPD PN and diabetic neuropathy share similar pathological mechanisms in terms of the voltage-gated sodium channels that are affected [1, 74]. Lastly, an interesting case report by Chen et al. reported 500 mg of methylcobalamin injection into a peroneal nerve under ultrasound guidance relieved peroneal neuropathy as evidenced by improvement of muscle power [75]. It is important to note powerful RCTs and cohort studies are required to assess the validity and generalizability of this case report to PN in IPD patients since Chen et al. demonstrated the efficacy of ultrasound guided vitamin B12 injections in nonPD patient suffering from foot drop. [1, 75].
Taken together, there is a major dearth in the literature regarding treatments of PN in PD patients. Majority of the studies focus on tailored treatments towards diabetic neuropathy. The current animal models, genetic studies, and clinical trials probing various drugs are all within the realm of diabetic neuropathy and other neuropathies, revealing the underlying pathological mechanisms [69,70,71,72,73,74]. These investigations are the right steps in the right direction in terms of elucidating management plans that can alleviate PN and improve the quality of life in PD patients. Importantly, RCTs and systematic reviews probing holistic and interdisciplinary treatments for PN in IPD is urgently needed [76,77,78,79].
Conclusion
Peripheral neuropathy (PN) is most commonly caused by diabetes, or is idiopathic. Though PN and Parkinson’s disease (PD) are distinct disease processes, it has become increasingly apparent that they are comorbid conditions, often resulting in treatment changes when found alone. l-DOPA, a staple treatment of PD, has been implicated in causing PN in PD patients, and is often correlated to increased Hcy and MMA levels. Possible treatment options for PN in PD includes combining l-DOPA with lesser used COMT-Is. COMT-Is help decrease Hcy and MMA levels, which is correlated to lesser disease burden. Screening for homocysteine and vitamin B12 levels along with neurophysiological and clinical monitoring for neuropathy is also advised in patients with PD who are undergoing l-DOPA treatment [12]. Future studies should investigate the toxic effects of elevated Hcy and MMA levels in patients with PD along with viable treatment options that include vitamin B12 and folate therapy. Large scale studies are required in order to understand the role and efficacy of COMT-Is along with other IPD interventions in PN.
The status of PN in PD is of much concern in the current day. PD pathophysiology and treatment are directly linked to PN development, often confusing the etiology of the condition. It is of absolute importance to determine the timeline of PN symptom presentation. This is especially important when PN is comorbid in a patient with PD, as it plays a critical part in the management of the conditions. With l-DOPA being a common cause or source of exacerbation of PN in PD patients, alternative therapies must be employed in treating both conditions. Furthermore, with clinical criteria of PN lacking, largely relying on EMG and biopsy, the diagnosis of PN in PD often lags behind and is often mismanaged with typical therapies that otherwise will not benefit the patient. Thus, although PN and PD are very well managed when independent of each other, they are often not treated to their maximal efficacies when present together due to the lack of understanding of the comorbid conditions.
Looking forward, the area of research focusing on association of PN and PD, and its management, is expected to expand. With further understanding of the pathophysiology behind PD and PN individually, the overlapping areas concerning pharmacotherapeutics is expected to grow. Considering recent clinical studies have included under 100 subjects, large-scale studies are likely to give greater support or opposition for specific causes, such as l-DOPA therapy with or without COMT-Is. Hence, it is expected that such studies will have an emphasis on biological markers, including Hcy and MMA, and how they are related to treatment response, much like Lamberti et al. and Toth et al. Considering this, it should also be expected that the future of research in this area explore the role of B12 and folate as adjunctive therapies for PN in PD.
References
Zis P, Grünewald RA, Chaudhuri RK, Hadjivassiliou M (2017) Peripheral neuropathy in idiopathic Parkinson’s disease: a systematic review. J Neurol Sci 378:204–209
England JD, Asbury AK (2004) Peripheral neuropathy. Lancet 363:2151–2161
Simon C (2009) Peripheral neuropathy. InnovAIT 2(9):538–545
Wolfe GI, Baker NS, Amato AA, Jackson CE, Nations SP, Saperstein DS, Cha CH, Katz JS, Bryan WW, Barohn RJ (1999) Chronic cryptogenic sensory polyneuropathy: clinical and laboratory characteristics. Arch Neurol 56(5):540–547
Latov N (2002) Diagnosis of CIDP. Neurology 59(12):S2–S6
Notermans NC, Wokke JH, van der Graaf Y (1994) Chronic idiopathic axonal polyneuropathy: a five year follow up. J Neurol Neurosurg Psychiatry 57:1525–1527
Muller T, Van Laar T, Cornblath DR et al (2013) Peripheral neuropathy in Parkinson’s disease: levodopa exposure and implications for duodenal delivery. Parkinsonism Relat Disord 19:501–507
Rajabelly YA, Martey J (2013) Levodopa, vitamins, ageing and the neuropathy of Parkinson’s disease. J Neurol 260:2844–2848
Leonard DR, Farooqi MH, Myer S (2004) Restoration of sensation, reduced pain, and improved balance in subjects with diabetic peripheral neuropathy: a double-blind, randomized, placebo-controlled study with monochromatic near-infrared treatment. Diabetes Care 27:168–172
Merola A, Romagnolo A, Zibetti M, Bernardini A, Cocito D, Lopiano L (2016) Peripheral neuropathy associated with levodopa–carbidopa intestinal infusion: a long-term prospective assessment. Eur J Neurol 23:501–509
Melamed E (1979) Early-morning dystonia: a late side effect of long-term levodopa therapy in Parkinson’s disease. Arch Neurol 36:308–310
Ceravolo R, Cossu G, Bandettini M (2013) Neuropathy and levodopa in Parkinson’s disease: evidence from a multicenter study. Mov Disord 28:1391–1397
Sakakibara R, Tateno F, Kishi M, Tsuyusaki Y, Terada H, Inaoka T (2014) MIBG myocardial scintigraphy in pre-motor parkinson's disease: a review. Parkinsonism Relat Disord 20:267–273
Müller T, Jugel C, Muhlack S, Klostermann F (2014) Methyl group–donating vitamins elevate 3-O-methyldopa in patients with Parkinson disease. Clin Neuropharmacol 36:52–54
Brattstrom L, Wilcken DEL (2000) Homocysteine and cardiovascular disease: cause or effect? Am J Clin Nutr 72:315–323
Jakubowski H (2006) Pathophysiological consequences of homocysteine excess. J Nutr 136:1741S–1749S
Mattson MP, Shea TB (2003) Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci 26:137–146
Rosenquist TH, Ratashak SA, Selhub J (1996) Homocysteine induces congenital defects of the heart and neural tube: effect of folic acid. Proc Natl Acad Sci U S A 93:15227–15232
Reynolds EH (2017) The risks of folic acid to the nervous system in vitamin B12 deficiency: rediscovered in the era of folic acid fortification policies. J Neurol Neurosurg Psychiatry 88:1097–1098
Brito A, Grapov D, Fahrmann J, Harvey D, Green R, Miller JW, Fedosov SN, Shahab-Ferdows S, Hampel D, Pedersen TL, Fiehn O, Newman JW, Uauy R, Allen LH (2017) The human serum metabolome of vitamin B-12 deficiency and repletion, and associations with neurological function in elderly adults. J Nutr 147:1839–1849
Okuma Y, Hattori N, Mizuno Y (2003) Sensory neuropathy in autosomal recessive juvenile Parkinism (PARK 2). Parkinsonism Relat Disord 9:313–314
Abbruzzese G, Pigullo S, Schenone A, Bellone E, Marchese R, di Maria E, Benedetti L, Ciotti P, Nobbio L, Bonifati V, Ajmar F, Mandich P (2004) Does Parkin play a role in the peripheral nervous system? A family report. Mov Disord 19:978–981
Jugel C, Ehlen F, Taskin B, Marzinzik F, Müller T, Klostermann F (2013) Neuropathy in Parkinson’s disease patients with intestinal levodopa infusion versus oral drugs. PLoS One:e666–e639
Nissinen E, Nissinen H, Larjonmaa H, Väänänen A, Helkamaa T, Reenilä I, Rauhala P (2005) The COMT inhibitor, entacapone, reduces levodopa-induced elevations in plasma homocysteine in healthy adult rats. J Neural Transm 112:1213–1221
Loens S, Chorbadzhieva E, Kleimann A, Dressler D, Schrader C (2017) Effects of levodopa/carbidopa intestinal gel versus oral levodopa/carbidopa on B vitamin levels and neuropathy. Brain Behav 7:e00698
Urban PP, Wellach I, Faiss S, Layer P, Rosenkranz T, Knop K, Weis J (2010) Subacute axonal neuropathy in Parkinson’s disease with cobalamin and vitamin B6 deficiency under duodopa therapy. Mov Disord 25:1748–1752
Meppelink AM, Nyman R, van Laar T, Drent M, Prins T, Leenders KL (2011) Transcutaneous port for continuous duodenal levodopa/carbidopa administration in Parkinson’s disease. Mov Disord 26(2):331–334
Antonini A, Isaias IU, Canesi M, Zibetti M, Mancini F, Manfredi L, Dal Fante M, Lopiano L, Pezzoli G (2007) Duodenal levodopa infusion for advanced Parkinson’s disease: 12- month treatment outcome. Mov Disord 22:1145–1149
Manca D, Cossu G, Murgia D, Molari A, Ferrigno P, Marcia E, Melis M (2009) Reversible encephalopathy and axonal neuropathy in Parkinson’s disease during duodopa therapy. Mov Disord 24:2293–2294
Galazky I, Schoof J, Stallforth S, Kupsch A, Heinze HJ, Kluge C (2014) Guillain–Barre/CIDP-like neuropathy in two Parkinsonian patients following intestinal levodopa/carbidopa treatment. Parkinsonism Relat Disord 20:125–127
Merola A, Zibetti M, Rizzone MG, Troiano M, Artusi CA, Angrisano S, Cocito D, Lopiano L (2014) Prospective assessment of peripheral neuropathy in Duodopa-treated parkinsonian patients. Acta Neurol Scand 129:e1–e5
Fernandez HH, Standaert DG, Hauser RA, Lang AE, Fung VS, Klostermann F, Lew MF, Odin P, Steiger M, Yakupov EZ, Chouinard S (2015) Levodopa-carbidopa intestinal gel in advanced Parkinson’s disease: final 12-month, open-label results. Mov Disord 30:500–509
Santos-García D, de la Fuente-Fernández R, Valldeoriola F, Palasí A, Carrillo F, Grande M, Mir P, De Fabregues O, Casanova J (2012) Polyneuropathy while on duodenal levodopa infusion in Parkinson’s disease patients: we must be alert. J Neurol 259:1668–1672
Rispoli V, Simioni V, Capone JG, Golfrè Andreasi N, Preda F, Sette E, Tugnoli V, Sensi M (2017) Peripheral neuropathy in 30 duodopa patients with vitamins B supplementation. Acta Neurol Scand 136:660–667
Devigili G, Rinaldo S, Lettieri C, Eleopra R (2016) Levodopa/carbidopa intestinal gel therapy for advanced Parkinson disease: AN early toxic effect for small nerve fibers? Muscle Nerve 54:970–972
Uncini A, Eleopra R, Onofrj M (2015) Polyneuropathy associated with duodenal infusion of levodopa in Parkinson’s disease: features, pathogenesis and management. J Neurol Neurosurg Psychiatry 86:490–495
Zoccolella S, Lamberti P, Armenise E, Mari M, Lamberti SV, Mastronardi R, Fraddosio A, Iliceto G, Livrea P (2005) Plasma homocysteine levels in Parkinson’s disease: role of antiparkinsonian medications. Parkinsonism Relat Disord 11:131–133
Goyette P, Pai A, Milos R, Frosst P, Tran P, Chen Z, Chan M, Rozen R (1998) Gene structure of human and mouse methylenetetrahydrofolate reductase (MTHFR). Mamm Genome 9:652–656
Gorgone G, Curro M, Ferlazzo N, Parisi G, Parnetti L, Belcastro V, Tambasco N, Rossi A, Pisani F, Calabresi P, Ientile R (2012) Coenzyme Q10, hyperhomocysteinemia and MTHFR C677T polymorphism in levodopa-treated Parkinson’s disease patients. NeuroMolecular Med 14:84–90
Schapira AH (2008) Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol 7:97–109
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK (2008) Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283:9089–9100
Wakabayashi K, Takahashi H, Takeda S, Ohama E, Ikuta F (1988) Parkinson’s disease: the presence of Lewy bodies in Auerbach's and Meissner's plexuses. Acta Neuropathol 76:217–221
Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J (2009 Jun 1) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634
Luoma P, Melberg A, Rinne JO (2004) Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet 364:875–882
Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T, Takahashi R, Hattori N, Imai Y, Lu B (2012) Parkinson’s disease – associated kinase PINK1 regulates axonal transport of mitochondria. PLoS Genet 8:e1002537
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55:181–184
Dabby R, Djaldetti R, Shahmurov M, Treves TA, Gabai B, Melamed E et al (2006) Skin biopsy for assessment of autonomic denervation in Parkinson's disease. J Neural Transm 113:e1169–e1176
Tolosa E, Wenning G, Poewe W (2006) The diagnosis of Parkinson’s disease. Lancet Neurol 5:75–86
Nolano M, Provitera V, Estraneo A, Selim MM, Caporaso G, Stancanelli A, Saltalamacchia AM, Lanzillo B, Santoro L (2008) Sensory deficit in Parkinson’s disease: evidence of a cutaneous denervation. Brain 131:1903–1911
Orimo S, Amino T, Yokochi M, Kojo T, Uchihara T, Takahashi A, Wakabayashi K, Takahashi H, Hattori N, Mizuno Y (2005) Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord 20:1350–1353
Orimo S, Oka T, Miura H, Tsuchiya K, Mori F, Wakabayashi K, Nagao T, Yokochi M (2002) Sympathetic cardiac denervation in Parkinson’s disease and pure autonomic failure but not in multiple system atrophy. J Neurol Neurosurg Psychiatry 73:776–777
Braak H, del Tredici K, Rüb U, De Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Del Tredici K, Rüb U, De Vos RA, Bohl JR, Braak H (2002) Where does Parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol 61:413–426
Orimo S, Takahashi A, Uchihara T, Mori F, Kakita A, Wakabayashi K, Takahashi H (2007) Degeneration of cardiac sympathetic nerve begins in the early disease process of Parkinson's disease. Brain Pathol 17:24–30
Donadio V, Incensi A, Leta V, Giannoccaro MP, Scaglione C, Martinelli P et al (2014) Skin nerve alpha-synuclein deposits: a biomarker for idiopathic Parkinson disease. Neurology 82:e1362–e1369
Fujishiro H, Frigerio R, Burnett M, Klos KJ, Josephs KA, Delledonne A et al (2008) Cardiac sympathetic denervation correlates with clinical and pathologic stages of Parkinson’s disease. Mov Disord 23:e1085–e1092
Lebouvier T, Neunlist M, Bruley des Varannes S, Coron E, Drouard A, N'Guyen JM et al (2010) Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS One 5:e127–e128
Doppler K, Ebert S, Uceyler N, Trenkwalder C, Ebentheuer J, Volkmann J et al (2014) Cutaneous neuropathy in Parkinson’s disease: a window into brain pathology. Acta Neuropathol 128:e99–e109
Wang N, Gibbons CH, Lafo J, Freeman R (2013) Alpha-synuclein in cutaneous autonomic nerves. Neurology 81:e1604–e1610
Donadio V, Incensi A, Piccinini C, Cortelli P, Giannoccaro MP, Baruzzi A, Liguori R (2016) Skin nerve misfolded α-synuclein in pure autonomic failure and Parkinson disease. Ann Neurol 79:306–316
Gondim Fde A, de Oliveira GR, Peixoto AA Jr, Horta WG (2010) A case series of peripheral neuropathy in patients with Parkinson’s disease. Ann Neurol 68:973–975
Rajabally YA, Martey J (2011) Neuropathy in Parkinson disease: prevalence and determinants. Neurology 77:e1947–e1950
Gusmaroli G, Barbagli D, Ravagnani M, Mongiovetti M, Capone L, Tarletti R et al (2010) Axonal polyneuropathy during duodenal levodopa treatment in a woman with idiopathic Parkinson’s disease. Mov Disord 25:S720
Palasí A, Fábregues O, Hernández-Vara J, Seoane JL, Gámez J, Álvarez-Sabín J. Small fiber peripheral neuropathy presentation after starting treatment with continuous intraduodenal levodopa infusion in one patient with Parkinson disease. In: Poster presentation at: 7th international congress on mental dysfunction and other non-motor features in Parkinson’s disease 2010
Klostermann F, Jugel C, Müller T, Marzinzik F (2012) Malnutritional neuropathy under intestinal levodopa infusion. J Neural Transm 119:e369–e372
Toth C, Brown MS, Furtado S, Suchowersky O, Zochodne D (2008) Neuropathy as a potential complication of levodopa use in Parkinson’s disease. Mov Disord 23:1850–1859
Shanb AA, Youssef EF, Al Baker WI, Al-Khamis FA, Hassan A, Jatoi NA (2020) The efficacy of adding electromagnetic therapy or laser therapy to medications in patients with diabetic peripheral neuropathy. J Lasers Med Sci 11(1):20–28
Cossu G, Ceravolo R, Zibetti M, Arca R, Ricchi V, Paribello A, Murgia D, Merola A, Romagnolo A, Nicoletti V, Palermo G (2016) Levodopa and neuropathy risk in patients with Parkinson disease: effect of COMT inhibition. Parkinsonism Relat Disord 27:81–84
Sun T et al (2005) Effectiveness of vitamin B12 on diabetic neuropathy: systematic review of clinical controlled trials. Acta Neurol Taiwan 14(2):48–54
Picci C, Wong VS, Costa CJ, McKinnon MC, Goldberg DC, Swift M, Alam NM, Prusky GT, Shen S, Kozikowski AP, Willis DE (2020) HDAC6 inhibition promotes α-tubulin acetylation and ameliorates CMT2A peripheral neuropathy in mice. Exp Neurol 5:113281
Lazic A, Popović J, Paunesku T, Woloschak GE, Stevanović M (2020) Insights into platinum-induced peripheral neuropathy–current perspective. Neural Regen Res 15(9):1623–1630
Massa F, Zuppa A, Pesce G, Demichelis C, Bergamaschi M, Garnero M, Briani C, Ferrari S, Schenone A, Benedetti L (2020) Bendamustine–rituximab (BR) combined therapy for treatment of immuno-mediated neuropathies associated with hematologic malignancy. J Neurol Sci 413:116777
Prnova MS, Kovacikova L, Svik K, Bezek S, Elmazoğlu Z, Karasu C, Stefek M (2019) Triglyceride-lowering effect of the aldose reductase inhibitor cemtirestat—another factor that may contribute to attenuation of symptoms of peripheral neuropathy in STZ-diabetic rats. Naunyn Schmiedeberg's Arch Pharmacol 5:1–1
Elkholy SE, Elaidy SM, El-Sherbeeny NA, Toraih EA, Elgawly HW (2020) Neuroprotective effects of ranolazine versus pioglitazone in experimental diabetic neuropathy: targeting Nav1. 7 channels and PPAR-γ. Life Sci 14:117557
Chen CH, Huang YK, Jaw FS (2015) Ultrasound-guided perineural vitamin b12 injection for peripheral neuropathy. J Med Ultrasound 23:104–106
Nardone A, Grasso M, Schieppati M (2006) Balance control in peripheral neuropathy: are patients equally unstable under static and dynamic conditions? Gait Posture 23:364–373
Hoeijmakers JG, Faber CG, Lauria G, Merkies IS, Waxman SG (2012) Small-fibre neuropathies – advances in diagnosis, pathophysiology and management. Nat Rev Neurol 8:369–379
Nyholm D (2012) Duodopa® treatment for advanced Parkinson’s disease: a review of efficacy and safety. Parkinsonism Relat Disord 18:916–929
Yeager D (2012) Diagnosing peripheral neuropathy. Aging Well 5:14
Acknowledgments
We would like to thank Zishan Chaudry, Uzma Ahmed, and Ali Ayub to their contributions to the manuscript.
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors report no conflicts of interest.
Ethical approval
The study protocol conforms to the provisions of the Declaration of Helsinki 1995, revised in Tokyo in 2004. We confirm that we have read the Journal’s position on ethical publication and affirm that this report is consistent with those guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dion A. Paul and Abdul Rehman M. Qureshi are co-first authors
Rights and permissions
About this article

Cite this article
Paul, D.A., Qureshi, A.R.M. & Rana, A.Q. Peripheral neuropathy in Parkinson’s disease. Neurol Sci 41, 2691–2701 (2020). https://doi.org/10.1007/s10072-020-04407-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-020-04407-4