
Abstract
NF-κB upregulation has been demonstrated in neurons and glial cells in response to experimental injury and neuropathological disorders, where it has been related to both neurodegenerative and neuroprotective activities. It has been generally recognized that NF-κB plays important roles in the regulation of apoptosis and inflammation as well as innate and adaptive immunity. However, the regulatory mechanism of NF-κB in apoptosis remained to be determined. The present study sought to first investigate the effect of a NF-κB inhibitor SN50, which inhibits NF-κB nuclear translocation, on cell death and behavioral deficits in our mice traumatic brain injury (TBI) models. Additionally, we tried to elucidate the possible mechanisms of the therapeutic effect of SN50 through NF-κB regulating apoptotic and inflammatory pathway in vivo. Encouragingly, the results showed that pretreatment with SN50 remarkably attenuated TBI-induced cell death (detected by PI labeling), cumulative loss of cells (detected by lesion volume), and motor and cognitive dysfunction (detected by motor test and Morris water maze). To analyze the mechanism of SN50 on cell apoptotic and inflammatory signaling pathway, we thus assessed expression levels of TNF-α, cathepsin B and caspase-3, Bid cleavage and cytochrome c release in SN50-pretreated groups compared with those in saline vehicle groups. The results imply that through NF-κB/TNF-α/cathepsin networks SN50 may contribute to TBI-induced extrinsic and intrinsic apoptosis, and inflammatory pathways, which partly determined the fate of injured cells in our TBI model.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI), a term applying to brain injury caused by external physical trauma, has long been thought to evoke primary neuronal degeneration in the site of injury and initiates a series of cellular events, including neuronal cell death, microglial activation, inflammation, and reactive astrogliosis, that lead to protracted secondary damage, impaired regeneration, and functional disabilities [1–4]. Two ways of neural cell death occur after TBI: neurons can die by necrosis caused by membrane disruption, irreversible metabolic disturbances and/or excitotoxicity immediately after mechanical trauma due to impact or penetration; or the other, neural cell death occurs in a delayed fashion, with morphological features of apoptosis [5]. Biochemical evidence of the reactivation of the apoptotic cascade after TBI both in experimental animals and in humans has been reported [5]. And it has been generally accepted that apoptosis can be segregated into two pathways, one involving the activation of a family of cysteine proteases termed ‘caspases’, and the other involving the release of apoptotic factors, like cytochrome, from mitochondria to cytosol [6].
NF-κB, a member of the Rel transcription factor family, plays important roles in the regulation of apoptosis and inflammation as well as innate and adaptive immunity [7–9]. Several studies have shown that activation of NF-κB in neurons has been generally involved in synaptic plasticity, neuronal function, development, and survival [7–10]; increased activation of NF-κB after brain injury has also been reported [4, 11, 12]. Besides, NF-κB activation plays a critical role in the onset of Hcy-induced apoptosis in neuroblastoma cells [13] and contributes to the QA-induced apoptosis of striatal neurons in vivo [14, 15], but its varying targets are far from being completely elucidated.
To delineate the role of NF-κB in TBI-induced cell apoptosis pathway in vivo, we studied the effect of inhibiting NF-κB activity on apoptosis in our controlled cortical impact model. SN50, a cell-permeable recombinant peptide to block NF-κB nuclear translocation [15, 16], has been widely used in the study of NF-κB transcription factor pathway [13–15]. Even the effect of SN50 has been evaluated in a corneal alkali burn model in mice [17], but this has never been referred in TBI models. Therefore, in the present study, we investigated the effect of SN50 on TBI-induced apoptosis and plasmalemma disruption of cortical or hippocampal cells and analyzed the possible mechanisms how NF-κB contributes to TBI-induced apoptosis.
In a mouse controlled cortical impact model, plasmalemma permeability to propidium iodide (PI) was an early and persistent feature of posttraumatic cellular injury [18]. In the present study, PI labeling was used to identify injured cells, and the numbers of PI-positive cells were counted. To explore whether PI-positive cells could represent TBI-induced cell loss, we assessed the loss of brain tissue after TBI. Furthermore, whether TBI-induced cell loss correlates with functional deficits was also investigated using motor test and Morris water maze. To explore the mechanism of NF-κB in TBI-induced cell death and functional outcome in our model, expression levels of proteins related to both extrinsic and intrinsic apoptotic pathways were tested in this study.
Materials and methods
Mouse traumatic brain injury model and drug administration
Mature male CD1 mice (20–25 g) were obtained from the Experimental Animal Center of Soochow University (certificate No 20020008, Grade II). The study was approved by the ethical committee of Soochow University. The NIH Guidelines for Care and Use of Laboratory Animals were followed in all animal procedures. Mice were anesthetized with 4 % chloral hydrate (0.4 mg/g) and positioned in a Kopf stereotaxic apparatus (Kopf, Tujunga, CA, USA) as described by Qin et al. [19]. A skin incision was performed to expose the skull and a 3-mm craniotomy was made lateral to the sagittal suture and centered between bregma and lambda using a dental drill. The bone flap was carefully removed without disruption of the underlying dura, and mice were subjected to TBI using a controlled cortical impact device [20, 21]. Following Feeney’s weight-drop model [21], a standardized parietal contusion was reproduced by letting a steel rod of 20 mg with a flat end and diameter of 2 mm drop on a piston resting on the dura from a height of 50 cm. The piston was allowed to compress the brain tissue at a depth of 1.0 mm. The reproducibility and consistency of this TBI model are determined by the accurate location, controlled hit weight, height, and duration. NF-κB inhibitor SN50 (total volume: 1 μl, concentration: 0.1 μg/μl, Sigma, St. Louis, MO, USA) or vehicle (normal saline, total volume: 1 μl) was administered into the ipsilateral cerebral ventricle (coordinates 1 mm posterior, 1 mm lateral, 2.5 mm deep to bregma) 10 min before TBI. After TBI, the bone flap was replaced, the scalp incision was sutured, and then mice were allowed to awaken and returned to their cages.
Assessment of propidium iodide (PI) staining
To observe the effect of SN50 on TBI-induced cell death, mice (n = 5/time point) were pretreated with SN50 or vehicle as described above, and killed at 1, 6, 12, 24, 48, and 72 h after operation. Loss of plasmalemma integrity was evaluated by intraperitoneal injection of PI (total volume: 100 μl, concentration: 0.4 mg/ml, Sigma, St. Louis, MO, USA) 1 h before killing the animal. The brain tissue was removed taking care to keep the contusion region intact, snap frozen in liquid nitrogen vapor, and then coronally sectioned at 12 μm thickness with cryostat (CM1900, Leica, Bensheim, Germany). A series of sections were collected 150–200 μm apart from anterior to posterior hippocampus from each brain (bregma −1.90 to −3.00). Cryostat sections were placed on poly-l-lysine coated glass slides and stored at −20 °C. Sections were fixed with 100 % alcohol for 15 min, and then 5–8 visual fields (200×) were randomly chosen from 5–8 sections of each brain to count PI-positive cells using a stereology microscope (Ludl Electronic Product Ltd). The mean number of positive cells for a given time point was calculated by summing the cell count data from all of the brain sections counted and dividing by the number of 200× fields analyzed [18].
Lesion volume measurements
To evaluate the effect of SN50 on TBI-induced cell loss, mice (n = 10/group) were pretreated with SN50 or vehicle as described above and killed 8 days after TBI. The brains were removed, post-fixed for 12 h, and then cryoprotected in 15 % sucrose in PBS. Frozen brain sections cut on a cryostat (25 μm) were collected at 500 μm sections and placed on poly-l-lysine coated glass slides, air dried, and stained with cresyl violet. Tissue damage was determined by morphometric image analysis. The areas of the lesion, injured and non-injured hemisphere and cortex were determined using an image analysis system (MCID, Imaging Research, St. Catherines, ON, Canada). Area measurements from each tissue section were obtained and summed, and corresponding volumes were calculated. Lesion volume was expressed in mm3 in the injured hemisphere and as percent volume of the non-injured hemisphere [22]. Lesion volume was quantitatively analyzed with Sigma Scan Pro 5. Average lesion volume in mm3 for each treatment group was calculated and a t test performed to determine significance of reduction in lesion volume compared with vehicle animals.
Assessment of motor function and spatial memory acquisition (Morris water maze)
Motor test was performed as described in the previous study [22]. Mice were gently grasped by the tail and placed on a 35-cm taut wire suspended between two upright wooden bars 50 cm above a padded table. Mice were held up to the wire so that they grip it with both front paws, ensuring that each mouse had equal chance to grasp the wire. The tail was carefully released. The length of time the mouse is able to remain on the wire (from 0 to 60 s) and the manner in which it holds on (one to four paws, tail, paws plus tail) is scored. A wire grip score is assigned each mouse based on the average of three trials per day.
To elucidate the effect of SN50 preconditioning against TBI-induced deficiency of spatial memory acquisition, Morris water maze experiment for mice was performed as described previously [22, 23]. Briefly, a circular pool 90 cm in diameter and 60 cm deep filled with 25 °C water to a depth of 27 cm was situated in a room with several highly visible cues located on the walls. A round, clear, plexiglass 10 cm diameter platform submerged 1 cm below the water level was positioned 15 cm from the wall of the goal quadrant. A video tracking system mounted above the pool (Chromotrack 3.0; San Diego Instruments, San Diego, CA, USA) recorded the swimming movements of the mice. Testing consists of a series of four trials/day for each mouse. For each trial, mice were randomized to one of the four starting locations (N, S, E, W) and placed in the pool facing the wall, allowing 90 s to find the goal and 15 s once the goal was achieved. If the mouse failed to find the goal within a 90-s period, it would be placed on the platform for 30 s. After one trial, mice were placed in a warmed chamber for 4 min. Performance of the mice in Morris water maze was quantified by the latency to find the platform and also the rate of learning over the trial period, expressed as change in latency per day, since this was shown to be more sensitive in detecting injury effects in mice. Latencies of trials were recorded using a video camera and analyzed with a tracking device and software (Chromotrack 3.0, San Diego Instruments), and results of all four trials each day for each mouse were averaged with the results of mice in the same treatment group.
Western blot analyses
To obtain accurate data for change in protein levels after TBI, animals (n = 5/time point) were pretreated with SN50 or vehicle and killed at designated times (1, 6, 12, 24 and 48 h) after TBI. The injured cortical or hippocampal region was dissected, and western blot analysis was performed as described by Qin et al. [18]. Tissue samples were homogenized in western blot lysis buffer containing 10 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 % (v/v) Triton X-100, 1 % sodium deoxycholate, 0.1 % SDS, 5 mM EDTA, 1 mM PMSF, 0.28 ku/l aprotinin, 50 mg/l leupeptin, 1 mM benzamidine, and 7 mg/l pepstatin A (all chemicals from Sigma-Aldrich, St. Louis, MO, USA). The homogenate was then centrifuged at 12,000 rpm for 10 min at 4 °C, and the protein level in the supernatants was determined using a Micro BCA protein assay kit with PBS as a blank standard (Pierce Chemical, Rockford, IL, USA). Aliquots containing 20 µg of protein were boiled in loading buffer containing 150 mM Tris (pH 6.8), 3 mM DTT, 6 % SDS, 0.3 % bromophenol blue and 30 % glycerol, and then were subject to electrophoresis on 10 % SDS-PAGE gel using a constant current [24]. Proteins were transferred to nitrocellulose membranes on a semi-dry electro transferring unit (Bio-Rad, California, USA) after electrophoresis and incubated with primary antibody against NF-κB p65 (Santa Cruz Biotechnology), TNF-α (Santa Cruz Biotechnology), caspase-3 (Santa Cruz Biotechnology), cathepsin B (Santa Cruz Biotechnology), tBid (Santa Cruz Biotechnology), cytochrome-c (monoclonal; BD PharMingen, San Diego, CA), HSP-60 (monoclonal; BD PharMingen), COX (polyclonal; Santa Cruz Biotechnology), in Tris-buffered saline containing 0.1 % Tween-20 (TBST), and 5 % non-fat dry milk for 3 h, followed by incubation with horseradish peroxidase-conjugated secondary antibody, respectively (anti-mouse, anti-rabbit or anti-goat IgG) in TBST for 1 h. Immunoreactivity was detected with enhanced chemiluminescent autoradiography (ECL kit, Amersham, Arlington Heights, IL, USA).
The result of the western blot analyses was scanned, and the signal intensity of primary antibody binding was quantitatively analyzed with Sigma Scan Pro 5 and was normalized to a loading control β-actin (mouse monoclonal antibody; Sigma).
Preparation of mitochondrial and cytosolic fractions
Isolation of mitochondria was performed as described by Qin et al. [24]. Mice were pretreated with ipsilateral cerebral ventricle (icv) infusion of SN50 (0.1 μg/μl) or vehicle (normal saline 1 μl) 10 min before TBI and were euthanized at 12, 24, and 48 h after TBI. Each injured cortical tissue (2 × 2 × 2 mm3 tissue block including the impact site and surroundings) was homogenized in 0.3 ml buffer A containing 250 mM sucrose, 1 mM EDTA, 50 mM Tris–HCl, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 0.28 U/ml aprotinin, 50 μg/ml leupeptin, and 7 μg/ml pepstatin A (pH 7.4) and homogenized with a glass Pyrex microhomogenizer (30 strokes). The homogenate was centrifuged at 1,000 rpm at 4 °C for 10 min, and then the resultant supernatant was transferred to a new tube to be centrifuged at 12,000 rpm at 4 °C for 20 min to obtain the mitochondrial pellet and supernatant that was then centrifuged at 100,000 rpm for 1 h at 4 °C to generate the cytosolic fraction. The mitochondrial pellet was washed three times in buffer B containing 250 mM sucrose, 1 mM EGTA, 10 mM Tris–HCl (pH 7.4), spun at 12,000 rpm at 4 °C for 10 min, and then lysed in western blot lysis buffer.
Statistics analysis
The data shown here represent the means ± SEM. Between-group differences in Western blot, Motor Test, PI-positive cells, Lesion volume, and Morris water maze data were analyzed using analysis of variance (ANOVA) with Dunnett t test post hoc tests in order to elucidate the effects of the vehicle- and SN50-treated groups against TBI. Statistical analysis was given using the related programs in SPSS 13.0. Statistical significance was considered at P < 0.05.
Results
Inhibition of NF-κB transcription factor pathway reduced TbI-induced cell loss
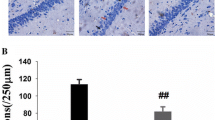
Previous studies reported that PI-positive cells were found in all groups (from 1 to 72 h) and peaked in 48 h time-point group in our TBI model [22, 25]. In this study, the number of PI-positive cells also peaked at 48 h and then declined in vehicle-treated groups (Fig. 1a–d). Pretreatment with SN50 resulted in a significant reduction in amount of PI-positive cells at 12, 24, and 48 h time-point post TBI compared with vehicle-treated groups (P < 0.05; Fig. 1e–h, I).

Time course of TBI-induced up-regulation of PI-positive cells. Mice were pretreated with intraperitoneal injection of PI 1 h before killing, and killed at 1, 6, 12, 24, 48, and 72 h after TBI treatment. Ten series of 12-μm sections (200 μm apart from each section) from anterior to posterior hippocampus was made from each animal brain. Frozen brain sections were fixed with 100 % alcohol for 15 min, and then five visual fields (×200) around the injured site were chosen to count PI-positive cells with stereology microscope. PI-positive cells were found in all experimental groups, and SN50 remarkably attenuated TBI-induced cell death at 12-, 24-, and 48-h time-point after injury. *P < 0.05 versus saline vehicle group at the same time point (n = 5). a–d PI-positive cells at 6, 12, 24, and 48 h in the saline-treated group; e–h PI-positive cells at 6, 12, 24, and 48 h in the SN50-treated group. Scale bars 200 μm
To determine whether PI-positive cells represent TBI-induced cell loss, the cumulative loss of brain tissue 8 days after TBI was also assessed (Fig. 2a). The results showed that TBI caused profound tissue loss of the brain, but pretreatment of SN50 significantly reduced the lesion volume compared with the vehicle-treated groups (Fig. 2b).

Effects of SN50 on TBI-induced lesions volume. a Eight days after TBI, all animals were killed. Brain sections obtained at 500 μm intervals spanning the length of the brain were stained with cresyl violet. b The areas of the lesion, injured and non-injured hemispheres, and cortex were determined using an image analysis system. Area measurements from each tissue section were obtained and summed, and corresponding volumes were calculated. Lesion volume was expressed in cubic millimeter in the injured hemisphere, and as percentage volume of the non-injured hemisphere. Lesion volume was quantitatively analyzed with Sigma Scan Pro 5. Average lesion volume in microliters for each treatment group was calculated and a t test (n = 10) performed to determine significance of reduction in lesion volume compared with saline vehicle animals. *P < 0.05 versus saline-treated group. Note the injured hemisphere (arrows)
Inhibition of NF-κB transcription factor pathway reduced TBI-induced behavioral deficits
In the present study, TBI elicited a significant decline in motor performance from day 1 to day 4, which returned to nearly basal levels (5′) on the 5th day post injury. Treatment with SN50 accelerated the recovery of motor functional outcome (Fig. 3a) from 1st to 4th day.

Effects of SN50 on TBI-induced motor and cognitive deficits. a Motor function test was performed 1 day after injury. After TBI, motor impairment was observed as revealed by decreases in motor performance. SN50 reduced motor function deficits. b Learning and memory function test was performed 5 days after injury by Morris water maze. Mice were randomly assigned to either drug or vehicle groups. Animals (n = 20) subjected to pretreatment with SN50 demonstrated a significant decrease of visuospatial learning latencies relative to the saline group (P < 0.05) started at 7 days after TBI. *P < 0.05, # P < 0.01 versus saline vehicle group at the same time point
All TBI-treated animals displayed increased latencies in the ability to learn a visuospatial task that involved finding a platform after injury compared with that in the saline vehicle group (Fig. 3b), indicating that a cognitive dysfunction was induced by TBI. Animals subjected to SN50 pretreatment demonstrated a significant decrease in the visuospatial learning latencies relative to the saline vehicle group mice (P < 0.05) at 7 and 8 days post-TBI, indicating that SN50 treatment promotes cognitive functional recovery in our TBI model (Fig. 3b).
NF-κB transcription factor contributes to the extrinsic and the intrinsic apoptosis pathway
Change in NF-κB p65 protein levels
Up-regulation of NF-κB p65 was observed from 1 to 48 h in the injured cortex (Fig. 4a) and hippocampus (Fig. 4b) after TBI. Pretreatment with SN50 resulted in a significant reduction of NF-κB p65 protein levels from 6 to 48 h post-TBI (Fig. 4).

Reversal of TBI-induced increases in NF-κB by SN50. The time-course of TBI-induced activation of NF-κB in injured cortex (a) and hippocampus (b), and inhibition of TBI-induced activation of NF-κB by SN50. Optical densities of respective protein bands were analyzed with Sigma Scan Pro 5 and normalized with loading control (β-actin). Data were expressed as means ± SEM (n = 5). Statistical comparisons were carried out with ANOVA followed by Dunnett t test. *P < 0.05 and **P < 0.01 versus saline vehicle group at the same time point
Change in TNF-α protein levels
Up-regulation of TNF-α was observed from 1 to 48 h in the injured cortex (Fig. 5a) and at 48 h in the injured hippocampus (Fig. 5b) after TBI. Pretreatment with SN50 resulted in a significant reduction of TNF-α protein levels from 12 to 48 h post-TBI (Fig. 5).

Reversal of TBI-induced increases in TNF-α by SN50. The time-course of TBI-induced activation of TNF-α in injured cortex (a) and hippocampus (b), and inhibition of TBI-induced activation of TNF-α by SN50. Optical densities of respective protein bands were analyzed with Sigma Scan Pro 5 and normalized with loading control (β-actin). Data were expressed as means ± SEM (n = 5). Statistical comparisons were carried out with ANOVA followed by Dunnett t test. *P < 0.05 and **P < 0.01 versus saline vehicle group at the same time point
Changes in cathepsin B, truncated Bid (tBid), and cytochrome c protein levels
To examine the possible mechanism that inhibition of NF-κB by SN50 contributing to cell death in our TBI model, we examined the protein levels of cathepsin B, tBid and cytochrome c in SN50 pretreated group compared with those in vehicle saline group. The results showed that the protein levels of cathepsin B peaked at 24 h both in injured cortex and hippocampus after TBI and was significantly inhibited by administration of SN50 at 24 and 48 h post TBI (Fig. 6). Meanwhile, a significant increase in the amount of tBid was observed from 12 h and peaked at 24 h following TBI (Fig. 7). Upregulation of tBid was significantly blocked by pretreatment with SN50 at 24 and 48 h post TBI (Fig. 8). Furthermore, western blot analysis revealed that cytochrome c protein levels had a significant reduction in mitochondria and adversely a concomitant increase in the cytosol from 12 to 48 h after TBI (Fig. 8). Pretreatment with SN50 significantly reduced TBI-induced release of cytochrome c from mitochondria to cytosol at 24 and 48 h post TBI (Fig. 8).

Western blot analysis of cathepsin B in injured cortex (a) and hippocampus (b) after TBI. The time-course of TBI-induced up-regulation of cathepsin B, and reversal of TBI-induced increase in cathepsin B by SN50. The bars indicated the means ± SEM (n = 5). *P < 0.05 and **P < 0.01 versus saline group at the same time point

Inhibition of TBI-induced Bid cleavage by SN50 in injured cortex (a) and hippocampus (b). Animals were killed at various times after TBI as indicated. Extracts from the injured cortex and hippocampus were separated on SDS-PAGEL and protein levels of Bid were detected with immunoblotting. Optical densities of respective protein bands were analyzed with Sigma Scan Pro 5 and normalized with loading control (β-actin). Data were expressed as means ± SEM (n = 5). Statistical comparisons were carried out with ANOVA followed by Dunnett t test. *P < 0.05 versus saline group at the same time point

Attenuation of TBI-induced release of cytochrome c in injured cortex by SN50. Animals were killed at various times after TBI as indicated. a Cortical tissues were dissected for isolation of mitochondrial and cytosolic fractions. The cyt-c, COX protein levels in mitochondria and cytosol were determined with western blot analysis. b, c Optical densities of the protein bands were analyzed with Sigma Scan Pro 5 and normalized with loading control (HSP60 or β-actin). Bars represent means ± SEM (n = 5). Statistical analysis was carried out with ANOVA followed by Dunnett t test. *P < 0.05 versus saline vehicle group at the same time point; # P < 0.05 and ## P < 0.01 versus naive group
Change in caspase-3 protein levels
The results demonstrated that the protein levels of active form of caspase-3 (p20) had a modest increase from 12 to 48 h after TBI (Fig. 9). Blockage of NF-κB transcription factor pathway by SN50 significantly suppressed TBI-induced activation of caspase-3 from 12 to 48 h post TBI (Fig. 9).

Inhibition of TBI-induced caspase-3 activation by SN50. a The time-course of TBI-induced activation of caspase-3 and inhibition of TBI-induced activation of caspase-3 by SN50 in injured cortex and hippocampus. b Optical densities of respective protein bands were analyzed with Sigma Scan Pro 5 and normalized with loading control (β-actin). Data were expressed as means ± SEM (n = 5). Statistical comparisons were carried out with ANOVA followed by Dunnett t test. *P < 0.05 and **P < 0.01 versus saline vehicle group at the same time point
Discussion
Traumatic brain injury (TBI) induced cell death occurs during a period of days to months both in TBI model and patients [5, 26, 27]. Despite advances in understanding biochemical mechanisms that contribute to posttraumatic brain cell death, the time course of cell death remains to be characterized in experimental TBI models. PI permeability has been recognized as a marker for eventual cell death and PI labeling has been widely used to detect cell death in vivo [27, 28]. In this study, we found that, as our previous study, the number of PI-positive cells peaked at 48 h rather than at earlier time points post TBI, suggesting that secondary injury mechanisms contributed to the destruction of plasmalemma membrane integrity, thereby priming neuronal cell death.
Previous studies have reported that NF-κB was immediately increased in neuronal cells of the degenerating cortex as well as in astrocytes located in the corpus callosum adjacent to cortical trauma in the immature rat brain [4], which suggested an important role of NF-κB activation in the early mechanisms of neuronal death or survival, as well as in the development of the glial and inflammatory responses following traumatic injury to the immature rat brain. But whether NF-κB activation contributes to cell death or survival remains unclear. In the present study, we found that SN50, a cell-permeable peptide inhibitor of NF-κB, significantly suppressed plasmalemma disruption and cumulative loss of cells, demonstrated a better performance of motor function, as well as had a significant decrease in the visuospatial learning latencies in our mouse TBI model, suggesting that pretreatment with SN50 provided neuroprotection against cell death induced by subsequent cerebral TBI. In our mouse TBI model, we have previously reported that most of the PI-positive cells were neurons [22], which were considered to be associated with motor and cognition ability. This may be also proved in our present study: compared with saline-treated group, SN50-treated group presented less cell loss, demonstrated a better performance of motor function (detected by motor test), and had a significant decrease in the visuospatial learning latencies (detected by Morris water maze).
Previous studies have reported that plasmalemma disruption is mainly caused by the mechanical external force of TBI and the secondary injury mechanisms contributed to the destruction of plasmalemma membrane integrity, thereby priming programmed cell death of nerve cells [29]. The present study focused on the best understood apoptotic pathways. Caspase-dependent apoptosis can occur via extrinsic or intrinsic pathways: extrinsic pathways involve members of the death receptor superfamily including TNF-R1, while intrinsic apoptotic pathways are activated by stimuli involving mitochondrial membrane permeabilization and then release of proapoptotic mitochondrial proteins (cytochrome c, etc.) into the cytosol, leading to activation of caspases and cell death [6]. These two pathways can both lead to caspase-3 cleavage, whereupon the process of apoptosis is irreversible. In this study, we found that TBI-induced activation of TNF-α was significantly suppressed in the SN50-pretreated group compared with that in the vehicle-treated group, indicating that, through attenuating expression levels of TNF-α, SN50 may attenuate TNF-α-mediated extrinsic apoptotic pathway and inflammation response following TBI.
We also found that SN50 significantly suppressed activation of cathepsin B, cleavage of Bid, release of cytochrome c from mitochondria, and activation of caspase-3 at 24 and 48 h post TBI. It has been reported that relocalized lysosomal cathepsin B can process Bid to active tBid [3, 30, 31], and truncated Bid (tBid) translocates to mitochondrial membranes to cause cytochrome c and apoptosis-activating factor release from mitochondria, which triggers the inner apoptotic active cascade [32, 33]. Thus, we assume that SN50 may contribute to mitochondrial (intrinsic) apoptotic pathway by direct or indirect (through NF-кB pathway) regulation of cathepsin B. Moreover, NF-кB has been well known as an upstream regulator of inflammation which activates TBI-induced inflammatory cytokines like TNF-α. Several studies have reported that TNF-α cytotoxic signaling, as one of important members of the death receptor, involved lysosomal permeabilization with release of the lysosomal protease cathepsin B into the cytosol [34], and also involved in activation of NF-κB. Based on our data, NF-кB/TNF-α/cathepsin networks was, at least partly, involved in the mechanism of anti-apoptotic action of SN50 on TBI-induced cell death. This is consistent with previous studies which reported that SN50 prevented H2O2 induced-apoptosis in SH-SY5Y neural cells [35].
In conclusion, SN50 preconditioning, induced by administration of low-dose SN50, somewhat provides neuroprotection in the injured cerebral cortex and hippocampus that may finally lead to temporary or even permanent cognitive and motor dysfunction in our TBI models. The protective effects of SN50 preconditioning may be related with part suppression of apoptosis as well as reduction of inflammation cytokines like TNF-α. However, further study is needed to completely elucidate the protective mechanism of SN50 contributing to TBI-induced cell death and to establish its clinical utility in the treatment of TBI to offer a potent strategy for the development of therapeutic interventions for TBI.
References
Jain KK (2008) Neuroprotection in traumatic brain injury. Drug Discovery Today 13:23–24
Di-Giovanni S, Movsesvan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI (2005) Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci USA 23:8333–8335
Uryu K, Laurer H, McIntosh T, Pratico D, Martinez D, Leight S, Lee VM, Trojanowski JQ (2002) Repetitive mild brain trauma accelerates Aβ deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci 22:446–454
Sanz Olga, Acarin Laia, Gonza’lez Berta, Castellano Bernardo (2002) NF-κB and IκBα expression following traumatic brain injury to the immature rat brain. J Neurosci Res 67:772–780
Zhang X, Chen Y, Jenkins LW, Kochanek PM, Clark RSB (2005) Bench-to-bedside review: apoptosis/programmed cell death triggered by traumatic brain injury. Crit Care 9:66–75
Zhang X, Chen Y, Jenkins LW, Kochanek PM, Clark RSB (2005) Bench-to-bedside review: Apoptosis/programmed cell death triggered by traumatic brain injury. Crit Care 9(1):66–75
Niederberger Ellen, Geisslinger Gerd (2010) Analysis of NF-κB signaling pathways by proteomic approaches. Expert Rev Proteomics 7(2):189–203
O’Neill LAJ, Kaltschmidt C (1997) NF-κB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci 20:252–258
Acarin L, Gonza’lez B, Castellano B (2000) STAT3 and NFκB activation precedes glial reactivity in the excitotoxically injured young cortex but not in the corresponding distal thalamic nuclei. J Neuropathol Exp Neurol 59:151–163
Mattson MP, Meffert MK (2006) Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ 13:852–860
Nonaka M, Chen XH, Pierce JES, Leoni MJ, McIntosh TK, Wolf JA, Smith DH (1999) Prolonged activation of NF-κB following traumatic brain injury in rats. J Neurotrauma 16:1023–1034
Pan DS, Liu WG, Yang XF, Cao F (2007) Inhibitory effect of progesterone on inflammatory factors after experimental traumatic brain injury. Biomed Environ Sci 20(5):432–438
Ferlazzo Nadia, Condello Salvatore, Currò Monica, Parisi Giulia, Ientile Riccardo, Caccamo Daniela (2008) NF-kappaB activation is associated with homocysteine-induced injury in Neuro2a cells. BMC Neurosci 9:62
Nakai M, Qin ZH, Chen JF, Wang Y, Chase TN (2000) Kainic acid induced apoptosis in rat striatum is associated with nuclear factor-kappaB activation. J Neurochem 74:647–658
Qin ZH, Wang YM, Nakai M, Chase TN (1998) Nuclear factor-kappa B contributes to excitotoxin-induced apoptosis in rat striatum. Mol Pharmacol 53:33–42
Walker PA, Harting MT, Jimenez F, Shah SK, Pati S, Dash PK, Cox CS Jr (2010) Direct intrathecal implantation of mesenchymal stromal cells leads to enhanced neuroprotection via an NFkappaB-mediated increase in interleukin 6 (IL-6) production. Stem Cells Dev 19(6):867–876
Saika S, Miyamoto T, Yamanaka O, Kato T, Ohnishi Y, Flanders KC, Ikeda K, Nakajima Y, Kao WW-Y, Sato M, Muragaki Y, Ooshima A (2005) Therapeutic effect of topical administration of SN50, an inhibitor of nuclear factor-κB, in treatment of corneal alkali burns in mice. Am J Pathol 166(5):1393–1403
Whalen MJ, Dalkara T, You Z, Qiu J, Bermpohl D, Mehta N, Suter B, Bhide PG, Lo EH, Ericsson M, Moskowitz MA (2008) Acute plasmalemma permeability and protracted clearance of injured cells after controlled cortical impact in mice. J Cereb Blood Flow Metab 28:490–505
Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, Chase TN (1999) NF-κB nuclear translocation up-regulates c-Myc and p53 during N-methyl-d-aspartate receptor-mediated apoptosis. J Neurosci 19:4023–4033
Tao LY, Chen XP, Ding M (2003) The study on expression of caspase-1 after brain contusion of different severity in rats. J Forensic Med 19:4–7
Feeney DM, Boyeson MG, Linnn RT, Murray HM, Dail WG (1981) Responses to cortical injury: I. Methodology and local effects of contusion in the rat. Brain Res 211:67–77
Luo CL, Chen XP, Yang R, Sun YX, Li QQ, Bao HJ, Cao QQ, Ni H, Qin ZH, Tao LY (2010) Cathepsin B contributes to traumatic brain injury induced cell death through mitochondria-mediated apoptotic pathway. J Neurosci Res 88:2847–2858
Satchell MA, Zhang X, Kochanek PM, Dixon CE, Jenkins LW, Melick JA, Szabo C, Clark RS (2003) A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14-3-3gamma. J Neurochem 85:697–708
Qin ZH, Wang Y, Kikly KK, Sapp E, Kegel KB, Aronin N, DiFiglia M (2001) Pro-caspase-8 is predominantly localized in mitochondria and released into cytoplasm upon apoptotic stimulation. J Biol Chem 276:8079–8086
Luo Chengliang, Chen Xiping, Ni Hong, Li Qianqian, Yang Rui, Sun Yuxia, Zhu Guangyou, Tao Luyang (2010) Comparison of labeling methods and time course of traumatic brain injury-induced cell death in mice. Neural Regen Res 5(9):706–709
Raghupathi R (2004) Cell death mechanisms following traumatic brain injury. Brain Pathol 14:215–222
Stoica BA, Faden AI (2010) Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 7(1):3–12
Krysko DV, Vanden-Berghe T, Herdek D, Vandenabeele P (2008) Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods 44:205–221
Shaw NA (2002) The neurophysiology of concussion. Prog Neurobiol 67:281–344
Reiners JJ, Caruso JA, Mathieu P, Chelladurai B, Yin XM, Kessel D (2002) Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves Bid cleavage. Cell Death Differ 9:934–944
Tardy C, Codogno P, Autefage H, Levade T, Andrieu-Abadie N (2006) Lysosomes and lysosomal proteins in cancer cell death (new players of an old struggle). Biochim Biophys Acta 1765:101–112
Kim R, Emi M, Tanabe K (2006) Role of mitochondria as the gardens of cell death. Cancer Chemother Pharmacol 57:545–553
Werneburg NW, Guicciardi ME, Bronk SF, Gores GJ (2002) Tumor necrosis factor-α-associated lysosomal permeabilization is cathepsin B dependent. Am J Physiol Gastrointest Liver Physiol 283(4):947–956
Werneburg N, Guicciardi ME, Yin XM, Gores GJ (2004) TNF-α-mediated lysosomal permeabilization is FAN and caspase 8/Bid dependent. Am J Physiol Gastrointest Liver Physiol 287(2):436–443
Ubertia D, Carsanaa T, Francisconia S, Ferrari Toninellia G, Canonicob PL, Memoa M (2004) A novel mechanism for pergolide-induced neuroprotection: inhibition of NF-kB nuclear translocation. Biochem Pharmacol 67:1743–1750
Acknowledgments
The work was supported by the National Science Foundation of China (No. 30571909, 30872666, 30870808, 81172911) and the Shanghai Forensic Key Lab Foundation (No. KF0904, KF1005).
Author information
Authors and Affiliations
Corresponding author
Additional information
Y.-X. Sun and D.-K. Dai equally contributed to this work.
Rights and permissions
About this article
Cite this article
Sun, YX., Dai, DK., Liu, R. et al. Therapeutic effect of SN50, an inhibitor of nuclear factor-κB, in treatment of TBI in mice. Neurol Sci 34, 345–355 (2013). https://doi.org/10.1007/s10072-012-1007-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-012-1007-z