
Abstract
Chronic anterior uveitis is the most frequent among extra-articular manifestations of juvenile idiopathic arthritis (JIA) and a relevant cause of ocular morbidity in children. Asymmetric arthritis, early onset disease, female sex, and anti-nuclear antibody (ANA) positivity are counted among risk factors for developing this complication. It usually has insidious onset and asymptomatic chronic-relapsing course, but the persistence of low-grade chronic inflammation can lead to irreversible structural ocular damage and to vision-threatening complications. For such reasons, achieving a complete absence of inflammation through early targeted and aggressive treatments is a primary therapeutic goal in these patients. This review is aimed at summarizing scientific evidence about biologic rescue therapy of JIA-related uveitis in patients who fail to achieve clinical remission, in spite of being treated with conventional disease-modifying anti-rheumatic drugs (cDMARDs) and at least one biologic tumor necrosis factor (TNF)-α inhibitor. Interleukin (IL)-6 inhibition appears a promising and safe option for refractory JIA-related uveitis. Abatacept and rituximab proved to be beneficial as well, but their efficacy together with some safety concerns needs to be more extensively evaluated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Uveitis is a well-known vision-threatening complication of juvenile idiopathic arthritis (JIA) and is considered the most frequent among the extra-articular manifestations of the disease, with a mean frequency of 12–16% and a remarkable geographic variability. JIA-extended oligoarthritis course type is traditionally counted among risk factors for developing chronic anterior uveitis [1, 2]. Nevertheless, it is quite accepted among experts that a group of patients currently categorized in different JIA subtypes (oligoarticular persistent, oligoarticular extended, polyarticular seronegative, and psoriatic) shares a cluster of clinical features that strongly suggest a common background and a high risk for developing chronic anterior uveitis during the disease course: asymmetric arthritis, early onset disease, female predominance, frequent anti-nuclear antibody (ANA) positivity, and association with HLA-DR8 [3]. In this context, uveitis can precede the onset of arthritis in 3–7% of cases, but most patients develop ocular disease concomitantly or within the first 4 years after the occurrence of arthritis [4]. Though several forms of uveitis have been reported in association with JIA, the most common presentation is bilateral non-granulomatous anterior uveitis, characterized by insidious onset and asymptomatic chronic-relapsing course, leading to irreversible structural ocular damage [5]. The persistence of low-grade underlying active inflammation, even if clinical signs are absent, is associated with an increased risk of developing visual impairment over time, owing to the occurrence of structural complications as band keratopathy, posterior synechiae, cataract, glaucoma, hypotony, macular edema, epiretinal membrane, and optic disk edema [6,7,8]. Hence, all patients in whom a diagnosis of JIA is being considered should be screened for uveitis within 4–6 weeks from referral and then at 3–12-month intervals depending on risk stratification [9] (Table 1), and immunosuppressive therapy should be initiated without any delay when ocular inflammation is detected.
This review is aimed at summarizing scientific evidence about biologic rescue therapy of JIA-U in patients who fail to achieve clinical remission, in spite of being treated with cDMARDs and at least one biologic TNF-α inhibitor.
Methods
An extensive literature search in the MEDLINE database (via PubMed) has been performed up to December 2018. The following words were searched in Medical Subject Headings: “arthritis, juvenile rheumatoid” and “uveitis” or a number of synonyms of the previous ones (“jia” or “jra” or “juvenile” or “child” or “pediatric” or “paediatric” and “rheumatoid” or “rheumatic” or “idiopathic” or “chronic” or “systemic” and “arthritis” or “arthritides” or “polyarthritis” or “oligoarthritis” or “still’s disease” or “still disease” and “paediatric” or “pediatric” or “child” or “juvenile” and “uveitis” or “iridocyclitis” or “vitritis” or “iritis” or “panuveitis” or “cyclitis” or “pan-uveitis”) were searched as entree terms as well. Observational studies, clinical trials, case series, and reviews were carefully screened for eligibility in relation to the purpose of our manuscript. Papers were included if the outcome of any biological rescue treatment, both in children and in adults diagnosed with JIA-U refractory to TNF-α inhibition, was reported. Papers written in languages other than English, published before 2001, or not providing data about the main focus of this research were excluded. On the contrary, given the lack of randomized clinical trials and prospective studies, single case reports and short case series have been included in the literature review.
Tocilizumab
Tocilizumab is a fully humanized antibody that binds both to soluble and membrane-bound interleukin-6 (IL-6) receptors, inhibiting its pro-inflammatory effects. Several studies documented high levels of this cytokine in the vitreous fluid [10], in the aqueous humor [11,12,13], and also in the sera [14] of patients affected by non-infectious uveitis, suggesting the opportunity of targeting the IL-6 pathway in order to treat different clinical conditions such as chronic idiopathic uveitis, Vogt-Koyanagi-Harada syndrome, sarcoidosis, Behçet disease, HLA-B27-associated uveitis, and birdshot chorioretinopathy. Furthermore, IL-6 levels measured in the serum of rats affected by monophasic experimental autoimmune uveitis seem to positively correlate with clinical activity and histopathologic assessment of the disease [15].
IL-6 is a pleiotropic cytokine secreted by different cell lineages and displays a broad range of effects: it induces fever and production of acute phase reactants; it stimulates the differentiation of B lymphocyte in plasma cells and the activation of CD8+ cells in cytotoxic T cells; it promotes CD4+ T helper cell shift towards the Th17 subset rather than the T regulatory one [16]. The latter function, in particular, seems to play a significant role in the early phase of uveitis, suggesting interesting therapeutic opportunities centered on the IL-6–IL-17 pathway suppression [15, 17].
Tocilizumab appears the most promising biologic rescue treatment for JIA-U. It is administered by intravenous route, and doses usually adopted are the following: 8 mg/kg (max 800 mg) at 4-week intervals for patients at or above 30 kg weight, 10 mg/kg at 4-week intervals for patients less than 30 kg weight. The subcutaneous route of administration (162 mg every other week, followed by an increase to every week based on clinical response) is also reported in the treatment of JIA-U, with variable efficacy [18,19,20]. Concerning the safety profile, autoimmune cytopenia, increase in serum aminotransferases, gastrointestinal disorders, dizziness and nausea, allergic reactions, and increased risk of infections have been reported [16, 19].
The first report dealing with efficacy of tocilizumab in the treatment of JIA-U was provided by Tappeiner in 2012: he wrote about three adults with chronic anterior uveitis associated with vision-threatening complications, refractory to systemic steroid therapy, cDMARDs, and TNF-α inhibitors. The efficacy of tocilizumab was evaluated during a 6–12-month follow-up period. Clinical remission of uveitis and improvement of best-corrected visual acuity (BCVA) were obtained in two patients, while the third one showed no favorable response to the treatment [21]. Later, the same scientific group evaluated 17 children and young adults with severe course of persisting or refractory uveitis, treated with the IL-6 inhibitor, reporting a complete response in terms of uveitis inactivity in 10 patients after a mean of 5.7 months of treatment; furthermore, systemic glucocorticoids or cDMARDs could be spared in seven patients [22]. The multicenter study conducted by Calvo-Rìo evaluated the largest cohort of patients affected by severe JIA-U administered with tocilizumab, after the failure of cDMARDs and one to five different biologic agents. The authors disclosed a favorable clinical response to the anti-IL-6 treatment in 19 out of 25 patients, who achieved remission of uveitis at a median follow-up of 12 months; as secondary outcomes, significant reduction of prednisone dose and improvement of BCVA were also attained; nine patients who showed uveitic macular edema at baseline showed a statistically significant decrease in central foveal thickness (CFT) measured by OCT at the 12-month evaluation [19]. The efficacy of the inhibition of IL-6 activity in the treatment of patients affected by JIA and uveitic macular edema was reported also in other case series, suggesting tocilizumab as a promising therapeutic option for this complication [22,23,24,25,26]. Similarly, Adàn published a case report of a secondary retinal vasoproliferative tumor in a 29-year-old female with a long history of JIA-U, refractory to methotrexate, infliximab, and adalimumab. After 12 administrations of tocilizumab, the ophthalmic examination revealed a distinct regression of the tumoral mass at OCT and a significant improvement of BCVA [27]. Further case reports and case series are available in the literature, and they mostly validate the previously presented evidences [20, 28,29,30,31,32] (Table 2).
Abatacept
Abatacept is a fully human soluble fusion protein composed of two domains: the extracellular portion of cytotoxic T lymphocyte antigen 4 (CTLA-4) and a modified FC domain of immunoglobulin G1 (IgG1). The first domain binds to the costimulatory signals CD80 or CD86 on antigen presenting cells, acting as a competitor of their natural binding molecule CD28 on T cells. This interference selectively inhibits T cell activation, with consequences on many downstream cytokine pathways involved in the pathogenesis of autoimmune diseases [33]. Although not completely clarified, the role of T lymphocytes in the development of endogenous uveitis and JIA has been extensively studied [34,35,36]. The blockade of CD28-CD80/86 costimulation in experimental models of induced autoimmune anterior uveitis and uveoretinitis interfered with effector T cell generation, inhibited TNF-α production, and finally prevented the development of the autoimmune ocular disease [37, 38].
Abatacept has been licensed for intravenous use in patients affected by rheumatoid arthritis, psoriatic arthritis, and polyarticular JIA refractory to TNF-α inhibitors in Europe. The suggested dosage is 10 mg/kg (max 1000 mg) at 0–2–4 weeks and every 4 weeks thereafter. Abatacept administration is generally safe and well tolerated [33], sometimes associated to mild to moderate adverse events, such as nasopharyngitis, upper respiratory tract infections, vomiting, pyrexia, or acute infusion reactions of single occurrence and not requiring premedication; nevertheless, severe infections have been reported with a lower frequency [39]. In regard with abatacept use in children with JIA-U, post-infusion headache, weight gain, skin reactions, anaphylaxis, oral mycosis, and arthritis flare in one patient have been specifically reported [29, 40, 41].
Since the first patient with a sustained recovery from active JIA-U on abatacept treatment was reported [42], analogue attempts were made in patients refractory to TNF-α inhibitors, with variable results in terms of clinical efficacy and steroid-sparing effect [19, 21, 23,24,25, 29, 30, 40,41,42,43,44,45,46] (Table 3). A multinational retrospective study conducted by Birolo collected 31 patients affected by severe JIA-U, comparing abatacept as first-line biologic therapy (N = 14) and as a rescue treatment (N = 17); the costimulation inhibitor allowed patients to be free from new uveitis flares for more than 6 months in 17 out of 31 cases, with no significative difference between the two study groups [40]. Another retrospective evaluation of abatacept’s efficacy was performed by the Multinational Interdisciplinary Working Group for Uveitis in Childhood (MIWGUC) in a cohort of 21 children with resistant JIA-U (and vision-threatening complications in 17 of them); patients underwent ophthalmological controls every 3 months for 1 year, and 11 of them (52.4%) were found on clinical remission during at least one follow-up visit. Nevertheless, a new exacerbation of the disease was detected in eight patients during the follow-up and, furthermore, attempts of tapering concomitant systemic and local treatments were not successful in most cases [46]. More favorable results had been attained otherwise by the Italian group led by Zulian in 2010 on seven children administered with abatacept because of their refractory sight-threatening chronic anterior JIA-U and prospectively evaluated: in all of them, a 2-degree decrease or disappearance of inflammation (anterior chamber cells) was obtained in the first 6 months of therapy. Four patients were able to withdraw or reduce by half systemic glucocorticoid therapy. No new structural complications were detected during a follow-up period of 7 to 11 months, while the pre-existing ones (band keratopathy, posterior synechiae, cataract, vitritis, posterior vitreal detachment, and cystoid macular edema) remained substantially stable [41]. Interestingly, in a small case series published by Elhai, the efficacy of abatacept on disease control was not compromised when the intervals between administrations were widened, respectively, to 6 and 7 weeks apart [44]. A clinical trial on abatacept safety profile and efficacy in non-infectious uveitis has been recently completed (ClinicalTrials.gov Identifier: NCT01279954).
Rituximab
Rituximab is a genetically engineered chimeric murine/human monoclonal antibody directed towards CD20 antigen of mature B cells and it was initially developed for the treatment of lymphoproliferative disorders. Its immune-modulating effect has been proven to be nevertheless beneficial in many autoimmune conditions, including rheumatoid arthritis, immune-mediated cytopenias, neurologic disorders, systemic lupus erythematosus, granulomatosis with polyangiitis, and anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis [47]. Recent studies on circulating blood mononuclear cell gene expression profile disclosed a significant upregulation of B cell receptor signaling pathways in patients with JIA affected by the persistent oligoarticular type, as the strong correlation of this clinical subset of JIA with ANA positivity clearly suggests [48]. B cells targeted by rituximab are marked for destruction through antibody-dependent cell-mediated cytotoxicity, complement-mediated lysis, growth inhibition, or direct induction of apoptosis [49]. CD20 antigen is expressed on the surface of mature B lymphocytes, but neither on plasma cells nor on B lineage precursor stem cells, preserving immunoglobulin levels and ensuring a transient depletion of the circulating B compartment, including self-reactive lymphocytes [50]. It modulates also T cell response, promoting the differentiation of T regulatory cells [51, 52], not only as a consequence of B cell depletion and shift of their antigen-presenting function to dendritic cells and macrophages, but also directly by targeting subsets of T cells that produce pro-inflammatory cytokines and express low levels of CD20 or a cross-reacting antigen [53].
Rituximab is administered by intravenous route and different posologic regimens are adopted, according to diverse drug registration dosage schemes: 375 mg/mq, 500 mg/mq, 600 mg/mq, 1000 mg, or 500 mg on days 1 and 15 and recall infusions, if necessary (Table 4). No adverse events associated with rituximab are reported in literature in patients affected by JIA-U; nevertheless, in studies carried out on patients with rheumatoid arthritis [58] and non-Hodgkin’s lymphoma [59], mild-to-moderate infusion-related reactions (IRR) and less frequently anaphylaxis have been experienced during the first infusion of rituximab; the probability of infusion-related adverse events is higher in patients affected by lymphoma than in those with rheumatoid arthritis and decreases in both groups during subsequent administrations of the drug. Anyway, in order to prevent and reduce the severity of IRR, methylprednisolone, paracetamol, and antihistamines are commonly given before rituximab administration [www.ema.europa.eu]. As concerns the risk of infectious complications, they have been reported as being low, due to the stability of mean serum immunoglobulin levels [59].
CD20 antigen has been targeted in small cohorts of patients with refractory JIA-U, showing interesting results [54,55,56]. Conversely, other anecdotal reports achieved less favorable outcomes [19, 20, 24]. A retrospective multicenter case series conducted by Heiligenhaus evaluated the clinical outcome of ten patients with severe JIA-U with vision-threatening complications and active arthritis, who underwent two infusions of rituximab 375 mg/m2, after the failure of topical and systemic glucocorticoids, cDMARDs, and at least one TNF-α inhibitor. Uveitis improved in seven patients affected by ANA+ oligoarthritis, who succeeded in tapering down topical glucocorticoids and cDMARDs during a follow-up period of 7–18 months. New uveitis relapses occurred in four of the seven responders after 6–9 months and were attributed to the restoration of peripheral blood CD20 cells, but were successfully suppressed through a recall infusion of rituximab in all but one case. Uveitis activity persisted after rituximab treatment in three patients, suggesting a major role of long lived plasma cells as predominant producers of the autoantibodies in that subset of patients [54]. Valuable results have been obtained by Miserocchi, who managed eight patients with JIA-U, treated with rituximab 1000 mg for a mean period of 12 months after failure of three different TNF-α inhibitors. At the last follow-up assessment, seven of them achieved complete control of intraocular inflammation and were on persistent clinical remission, having withdrawn or tapered down the concomitant cDMARDs [55]. The same group published another analog series of eight patients affected by longstanding and refractory JIA-U in which rituximab allowed maintaining uveitis inactivity for a longer follow-up (26–62 months), with a considerable sparing of concomitant immunosuppressant medications. It is worth mentioning that two patients discontinued rituximab after 29 and 26 months because of lack of efficacy on arthritis, and that they were switched to golimumab [56]. Other papers available in the medical literature [25, 29, 30, 42, 57] mention more or less successful courses of the anti-CD20 monoclonal antibody rituximab for the treatment of refractory JIA-U, as listed in Table 4.
Others
Daclizumab is a humanized IgG monoclonal antibody produced by recombinant DNA technology, which targets CD25, the α-chain of the IL-2 receptor, expressed on the surface of human T lymphocytes once activated by mitogens or antigens, blocking the proliferative signal of this cytokine. As evaluated in experimental models of autoimmune uveoretinitis in the monkey, it significantly inhibits the IL-2-driven expansion of activated T cells and subsequently the development of the autoimmune ocular disease [60]. Intravenous daclizumab has been considered a promising rescue treatment for refractory non-infectious uveitis in childhood, since it had beneficial effects both on ocular inflammation and visual acuity at low dosage (1 mg/kg at 2–8-week intervals) [61]. In a 2009 pilot study, it led to a two-step reduction of anterior chamber inflammation within 12 weeks in four out of six patients with JIA-U when administered at high dose (8 mg/kg at baseline, followed by 4 mg/kg at week 2 and 2 mg/kg every 4 weeks thereafter). Nevertheless, the authors expressed the need for more extensive studies focusing on the safety profile associated with high-dose protocols [62]. Other papers reported that intravenous or subcutaneous daclizumab could effectively control the activity of severe intermediate and posterior non-infectious uveitis in 67–80% of adult patients, with a considerable steroid-sparing effect [63, 64]. However, daclizumab has been discontinued by the manufacturer in 2018 due to severe safety concerns related to the central nervous system [www.aifa.gov.it/sites/default/files/Zinbryta_DHPC_ITA_12-03-2018].
A rising interest on the efficacy of interleukin (IL)-1 blockers in several different inflammatory ocular diseases [65] has encouraged their employment also in pediatric patients affected by refractory JIA-U. A young girl with severe JIA-U, cataract, and cystoid macular edema, refractory to anti-TNF-α and abatacept, was treated with the IL-1beta monoclonal antagonist canakinumab (2 mg/kg every 4 weeks by subcutaneous injection) and concomitantly with oral prednisone and methotrexate; a 12-month follow-up showed both ocular remission and visual acuity recovery, in association with successful withdrawal of co-treatments [66]. On the other hand, further case reports have mentioned the use of the IL-1 receptor antagonist anakinra in patients affected by JIA-U [19, 24, 54].
The pathogenetic role of the IL-23/IL-17 axis has been studied both in experimental models of non-infectious uveitis and in several autoimmune systemic or ocular disorders [67, 68]. As concerning JIA, experimental evidences support a direct correlation between the number of peripheral Th17 cells and disease activity, to a point that a higher Th17 level predicts a longer period to reach remission [69]. A clinical report of an effective treatment with ustekinumab, the monoclonal antibody directed against IL-12 and IL-23, in a child with uveitis and ANA+ HLA-B27+ juvenile psoriatic extended oligoarthritis has been recently published by Salek: the patient was affected by anterior and intermediate uveitis since the age of four and was treated with systemic glucocorticoids, methotrexate, infliximab, and adalimumab. The introduction of ustekinumab 45 mg every 3 weeks allowed control of intraocular inflammation [32]. No other data about ustekinumab in JIA-U are currently available in the medical literature.
Conclusive remarks
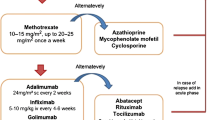
Consensus-based recommendations for the management of JIA-U have been recently developed by an experts’ group from the Single Hub and Access point for Pediatric Rheumatology in Europe (SHARE), with the purpose of overcoming the previous heterogeneity of clinical and scientific approach to anterior uveitis, a well-known vision-threatening complication occurring in children with JIA [70]. According to the recommendations, early introduction of methotrexate (MTX) is recommended if poor prognostic factors are present at the first evaluation or if uveitis inactivity is not reached in 3 months of topical glucocorticoids, to avoid the development of cataract and glaucoma [70]. In recent years, the importance of complete absence of inflammation as a primary therapeutic target in JIA patients has been extended also to the extra-articular manifestations of the disease [71]. Also, in regard of JIA-associated uveitis (JIA-U), the traditional step ladder treatment approach is being replaced by top-down algorithms, in which systemic management is started as soon as needed at the highest tolerated levels, in order to attain complete control of inflammation and to prevent or minimize the possibility of vision-threatening complications [8, 72]. Therefore, when conventional disease-modifying anti-rheumatic drugs (cDMARDs) fail to control ocular inflammation, switching or adding one tumor necrosis factor (TNF)-inhibitor agent is recommended [73]. On the basis of several cohort studies [74] and one randomized controlled trial [75], adalimumab is the biologic agent with the strongest scientific evidence of efficacy in JIA-U, when administered in association with MTX, infliximab is regarded as second choice, while etanercept should not be considered for these patients.
If the role of anti-TNF-α agents as first-line biologic drugs in patients with uveitis resistant to cDMARDs has reached a unanimous level of agreement among rheumatologists and ophthalmologists [73], variegate data are available in the medical literature about rescue solutions to manage anti-TNF-α failure in clinical practice. Experts’ opinion suggests testing anti-drug antibodies and drug trough level, and eventually increasing anti-TNF-α dosage or shortening intervals between administrations [76,77,78]. Furthermore, some data derived from descriptive studies suggest that also switching between different anti-TNF-α agents can be a reasonable strategy in order to achieve ocular remission [79, 80].
We have herein reviewed all scientific evidence about biologic drugs which can be considered as rescue therapy in managing children with severe refractory JIA-U. We noticed, even so, some critical issues, such as the lack of randomized controlled trials and comparative studies, the relatively small sample size of cohorts studied, and an overall heterogeneity of protocols and dosages employed, which make any effort to compare these results definitely unreliable. Taken into account the previously mentioned limitations, IL-6 inhibition appears a promising and safe option for these patients, also when vision-threatening macular edema complicates uveitis. In this regard, a multicenter phase II trial protocol of tocilizumab associated with methotrexate in anti-TNF-α refractory patients with JIA-U (the APTITUDE trial) is ongoing in the UK and is expected to end on April 2019 [https://doi.org/10.1186/ISRCTN95363507]. Abatacept proved to be beneficial in some study cohorts as well, and long-term outcome assessments would be desirable. Moreover, interesting data have been published on the anti-CD20 monoclonal antibody rituximab for resistant uveitis in JIA, but its efficacy together with some safety concerns related to the intravenous infusion of the drug need to be more extensively evaluated. A consistent effort to clarify the pathogenetic fundamentals of JIA-U and to identify reliable biomarkers of this challenging condition is necessary to identify higher-risk patients at an early stage of the disease and promptly address them to more-targeted and aggressive therapies.
References
Saurenmann RK, Levin AV, Feldman BM, Rose JB, Laxer RM, Schneider R, Silverman ED (2007) Prevalence, risk factors, and outcome of uveitis in juvenile idiopathic arthritis: a long-term follow-up study. Arthritis Rheum 56:647–657
Heiligenhaus A, Heinz C, Edelsten C, Kotaniemi K, Minden K (2013) Review for disease of the year: epidemiology of juvenile idiopathic arthritis and its associated uveitis: the probable risk factors. Ocul Immunol Inflamm 21:180–191
Martini A (2003) Are the number of joints involved or the presence of psoriasis still useful tools to identify homogeneous disease entities in juvenile idiopathic arthritis? J Rheumatol 30:1900–1903
Sen ES, Ramanan AV (2017) Juvenile idiopathic arthritis-associated uveitis. Best Pract Res Clin Rheumatol 31:517–534
Clarke SL, Sen ES, Ramanan AV (2016) Juvenile idiopathic arthritis-associated uveitis. Pediatr Rheumatol Online J 14:27
Paroli MP, Spinucci G, Fabiani C, Pivetti-Pezzi P (2010) Retinal complications of juvenile idiopathic arthritis-related uveitis: a microperimetry and optical coherence tomography study. Ocul Immunol Inflamm 18:54–59
Paroli MP, Fabiani C, Spinucci G, Abicca I, Sapia A, Spadea L (2013) Severe macular edema in patients with juvenile idiopathic arthritis-related uveitis. Case Rep Ophthalmol Med 2013:803989
Gregory AC, Kempen JH, Daniel E, Kaçmaz RO, Foster CS, Jabs DA, Levy-Clarke GA, Nussenblatt RB, Rosenbaum JT, Suhler EB, Thorne JE, Systemic Immunosuppressive Therapy for Eye Diseases Cohort Study Research Group (2013) Risk factors for loss of visual acuity among patients with uveitis associated with juvenile idiopathic arthritis: the systemic immunosuppressive therapy for eye diseases study. Ophthalmology 120:186–192
Bou R, Adán A, Borrás F, Bravo B, Calvo I, De Inocencio J, Díaz J, Escudero J, Fonollosa A, de Vicuña CG, Hernández V, Merino R, Peralta J, Rúa MJ, Tejada P, Antón J (2015) Clinical management algorithm of uveitis associated with juvenile idiopathic arthritis: interdisciplinary panel consensus. Rheumatol Int 35:777–785
Perez VL, Papaliodis GN, Chu D, Anzaar F, Christen W, Foster CS (2004) Elevated levels of interleukin 6 in the vitreous fluid of patients with pars planitis and posterior uveitis: the Massachusetts eye & ear experience and review of previous studies. Ocul Immunol Inflamm 12:193–201
El-Asrar AM, Struyf S, Kangave D, Al-Obeidan SS, Opdenakker G, Geboes K, Van Damme J (2011) Cytokine profiles in aqueous humor of patients with different clinical entities of endogenous uveitis. Clin Immunol 139:177–184
Yoshimura T, Sonoda KH, Ohguro N, Ohsugi Y, Ishibashi T, Cua DJ, Kobayashi T, Yoshida H, Yoshimura A (2009) Involvement of Th17 cells and the effect of anti-IL-6 therapy in autoimmune uveitis. Rheumatology 48:347–354
Kuiper JJ, Mutis T, de Jager W, de Groot-Mijnes JD, Rothova A (2011) Intraocular interleukin-17 and proinflammatory cytokines in HLA-A29-associated birdshot chorioretinopathy. Am J Ophthalmol 152:177–182.e1
Chen W, Zhao B, Jiang R, Zhang R, Wang Y, Wu H, Gordon L, Chen L (2015) Cytokine expression profile in aqueous humor and sera of patients with acute anterior uveitis. Curr Mol Med 15:543–549
Zhang L, Wan F, Song J, Tang K, Zheng F, Guo J, Guo D, Bi H (2016) Imbalance between Th17 cells and regulatory T cells during monophasic experimental autoimmune uveitis. Inflammation 39:113–122
Lopalco G, Fabiani C, Sota J, Lucherini OM, Tosi GM, Frediani B, Iannone F, Galeazzi M, Franceschini R, Rigante D, Cantarini L (2017) IL-6 blockade in the management of non-infectious uveitis. Clin Rheumatol 36:1459–1469
Hohki S, Ohguro N, Haruta H, Nakai K, Terabe F, Serada S, Fujimoto M, Nomura S, Kawahata H, Kishimoto T, Naka T (2010) Blockade of interleukin-6 signaling suppresses experimental autoimmune uveoretinitis by the inhibition of inflammatory Th17 responses. Exp Eye Res 91:162–170
Quesada-Masachs E, Caballero CM (2017) Subcutaneous tocilizumab may be less effective than intravenous tocilizumab in the treatment of juvenile idiopathic arthritis-associated uveitis. J Rheumatol 44:260–261
Calvo-Río V, Santos-Gómez M, Calvo I, González-Fernández MI, López-Montesinos B, Mesquida M, Adán A, Hernández MV, Maíz O, Atanes A, Bravo B, Modesto C, Díaz-Cordovés G, Palmou-Fontana N, Loricera J, González-Vela MC, Demetrio-Pablo R, Hernández JL, González-Gay MA, Blanco R (2017) Anti-Interleukin-6 receptor tocilizumab for severe juvenile idiopathic arthritis-associated uveitis refractory to anti-tumor necrosis factor therapy: a multicenter study of twenty-five patients. Arthritis Rheumatol 69:668–675
Curragh DS, O'Neill M, McAvoy CE, Rooney M, McLoone E (2018) Pediatric uveitis in a well-defined population: improved outcomes with immunosuppressive therapy. Ocul Immunol Inflamm 26:978–985
Tappeiner C, Heinz C, Ganser G, Heiligenhaus A (2012) Is tocilizumab an effective option for treatment of refractory uveitis associated with juvenile idiopathic arthritis? J Rheumatol 39:1294–1295
Tappeiner C, Mesquida M, Adán A, Anton J, Ramanan AV, Carreno E, Mackensen F, Kotaniemi K, de Boer JH, Bou R, de Vicuña CG, Heiligenhaus A (2016) Evidence for tocilizumab as a treatment option in refractory uveitis associated with juvenile idiopathic arthritis. J Rheumatol 43:2183–2188
Mesquida M, Molins B, Llorenç V, Sainz de la Maza M, Adán A (2014) Long-term effects of tocilizumab therapy for refractory uveitis-related macular edema. Ophthalmology 121:2380–2386
Mesquida M, Molins B, Llorenç V, Hernández MV, Espinosa G, Sainz de la Maza M, Adán A (2018) Twenty-four month follow-up of tocilizumab therapy for refractory uveitis-related macular edema. Retina 38:1361–1370
Adán A, Mesquida M, Llorenç V, Espinosa G, Molins B, Hernández MV, Pelegrín L (2013) Tocilizumab treatment for refractory uveitis-related cystoid macular edema. Graefes Arch Clin Exp Ophthalmol 251:2627–2632
Deuter CME, Zierhut M, Igney-Oertel A, Xenitidis T, Feidt A, Sobolewska B, Stuebiger N, Doycheva D (2017) Tocilizumab in Uveitic macular edema refractory to previous immunomodulatory treatment. Ocul Immunol Inflamm 25:215–220
Adán A, Mesquida M, Llorenç V, Modesto C (2014) Tocilizumab for retinal vasoproliferative tumor secondary to juvenile idiopathic arthritis-associated uveitis: a case report. Graefes Arch Clin Exp Ophthalmol 252:163–164
Silpa-Archa S, Oray M, Preble JM, Foster CS (2016) Outcome of tocilizumab treatment in refractory ocular inflammatory diseases. Acta Ophthalmol 94:e400–e406
Tsang AC, Roth J, Gottlieb C (2014) Tocilizumab for severe chronic anterior uveitis associated with juvenile idiopathic arthritis in a pediatric patient. Ocul Immunol Inflamm 22:155–157
Breitbach M, Tappeiner C, Böhm MR, Zurek-Imhoff B, Heinz C, Thanos S, Ganser G, Heiligenhaus A (2017) Discontinuation of long-term adalimumab treatment in patients with juvenile idiopathic arthritis-associated uveitis. Graefes Arch Clin Exp Ophthalmol 255:171–177
Souto FMS, Giampietro BV, Takiuti JT, Campos LMA, Hirata CE, Yamamoto JH (2018) Clinical features of paediatric uveitis at a tertiary referral Centre in São Paulo, SP, Brazil. Br J Ophthalmol. https://doi.org/10.1136/bjophthalmol-2018-312313
Salek SS, Pradeep A, Guly C, Ramanan AV, Rosenbaum JT (2018) Uveitis and juvenile psoriatic arthritis or psoriasis. Am J Ophthalmol 185:68–74
Ruperto N, Lovell DJ, Quartier P, Paz E, Rubio-Pérez N, Silva CA, Abud-Mendoza C, Burgos-Vargas R, Gerloni V, Melo-Gomes JA, Saad-Magalhães C, Sztajnbok F, Goldenstein-Schainberg C, Scheinberg M, Calvo Penades I, Fischbach M, Orozco J, Hashkes PJ, Hom C, Jung L, Lepore L, Oliveira S, Wallace CA, Sigal LH, Block AJ, Covucci A, Martini A, Giannini EH, for the Paediatric Rheumatology International Trials Organization (PRINTO) and the Pediatric Rheumatology Collaborative Study Group (PRCSG) (2008) Abatacept in children with juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled withdrawal trial. Lancet 372:383–391
Willermain F, Rosenbaum JT, Bodaghi B, Rosenzweig HL, Childers S, Behrend T, Wildner G, Dick AD (2012) Interplay between innate and adaptive immunity in the development of non-infectious uveitis. Prog Retin Eye Res 31:182–194
Caspi R (2008) Autoimmunity in the immune privileged eye: pathogenic and regulatory T cells. Immunol Res 42:41–50
Lin YT, Wang CT, Gershwin ME, Chiang BL (2011) The pathogenesis of oligoarticular/ polyarticular vs systemic juvenile idiopathic arthritis. Autoimmun Rev 10:482–489
Shao H, Woon MD, Nakamura S, Sohn JH, Morton PA, Bora NS, Kaplan HJ (2001) Requirement of B7-mediated costimulation in the induction of experimental autoimmune anterior uveitis. Invest Ophthalmol Vis Sci 42:2016–2021
Silver PB, Hathcock KS, Chan CC, Wiggert B, Caspi RR (2000) Blockade of costimulation through B7/CD28 inhibits experimental autoimmune uveoretinitis, but does not induce long-term tolerance. J Immunol 165:5041–5047
Ruperto N, Lovell DJ, Quartier P, Paz E, Rubio-Pérez N, Silva CA, Abud-Mendoza C, Burgos-Vargas R, Gerloni V, Melo-Gomes JA, Saad-Magalhães C, Chavez-Corrales J, Huemer C, Kivitz A, Blanco FJ, Foeldvari I, Hofer M, Horneff G, Huppertz HI, Job-Deslandre C, Loy A, Minden K, Punaro M, Nunez AF, Sigal LH, Block AJ, Nys M, Martini A, Giannini EH, Paediatric Rheumatology International Trials Organization and the Pediatric Rheumatology Collaborative Study Group (2010) Long-term safety and efficacy of abatacept in children with juvenile idiopathic arthritis. Arthritis Rheum 62:1792–1802
Birolo C, Zannin ME, Arsenyeva S, Cimaz R, Miserocchi E, Dubko M, Deslandre CJ, Falcini F, Alessio M, La Torre F, Denisova E, Martini G, Nikishina I, Zulian F (2016) Comparable efficacy of Abatacept used as first-line or second-line biological agent for severe juvenile idiopathic arthritis-related uveitis. J Rheumatol 43:2068–2073
Zulian F, Balzarin M, Falcini F, Martini G, Alessio M, Cimaz R, Cimino L, Zannin ME (2010) Abatacept for severe anti-tumor necrosis factor α refractory juvenile idiopathic arthritis-related uveitis. Arthritis Care Res (Hoboken) 62:821–825
Angeles-Han S, Flynn T, Lehman T (2008) Abatacept for refractory juvenile idiopathic arthritis-associated uveitis- a case report. J Rheumatol 35:1897–1898
Alpigiani MG, Salvati P, Vannati M, Callegari S, Trovato F, Rossi R, Gamba S, Lorini R (2010) Abatacept for severe anti TNF-alpha refractory JIA-associated uveitis: a case report. 17th Pediatric Rheumatology European Society Congress. September 9-12 València, Spain
Elhai M, Deslandre CJ, Kahan A (2011) Abatacept for refractory juvenile idiopathic arthritis-associated uveitis: two new cases. Comment on the article by Zulian et al. Arthritis Care Res (Hoboken) 63:307–308 author reply 308
Kenawy N, Cleary G, Mewar D, Beare N, Chandna A, Pearce I (2011) Abatacept: a potential therapy in refractory cases of juvenile idiopathic arthritis-associated uveitis. Graefes Arch Clin Exp Ophthalmol 249:297–300
Tappeiner C, Miserocchi E, Bodaghi B, Kotaniemi K, Mackensen F, Gerloni V, Quartier P, Lutz T, Heiligenhaus A (2015) Abatacept in the treatment of severe, longstanding, and refractory uveitis associated with juvenile idiopathic arthritis. J Rheumatol 42:706–711
MacIsaac J, Siddiqui R, Jamula E, Li N, Baker S, Webert KE, Evanovitch D, Heddle NM, Arnold DM (2018) Systematic review of rituximab for autoimmune diseases: a potential alternative to intravenous immune globulin. Transfusion 58:2729–2735
Barnes MG, Grom AA, Thompson SD, Griffin TA, Pavlidis P, Itert L, Fall N, Sowders DP, Hinze CH, Aronow BJ, Luyrink LK, Srivastava S, Ilowite NT, Gottlieb BS, Olson JC, Sherry DD, Glass DN, Colbert RA (2009) Subtype-specific peripheral blood gene expression profiles in recent-onset juvenile idiopathic arthritis. Arthritis Rheum 60:2102–2112
Kazkaz H, Isenberg D (2004) Anti B cell therapy (rituximab) in the treatment of autoimmune diseases. Curr Opin Pharmacol 4:398–402
Miserocchi E, Modorati G (2012) Rituximab for noninfectious uveitis. Dev Ophthalmol 51:98–109
Sfikakis PP, Souliotis VL, Fragiadaki KG, Moutsopoulos HM, Boletis JN, Theofilopoulos AN (2007) Increased expression of the FoxP3 functional marker of regulatory T cells following B cell depletion with rituximab in patients with lupus nephritis. Clin Immunol 123:66–73
Catzola V, Battaglia A, Buzzonetti A, Fossati M, Scuderi F, Fattorossi A, Evoli A (2013) Changes in regulatory T cells after rituximab in two patients with refractory myasthenia gravis. J Neurol 260:2163–2165
Hultin LE, Hausner MA, Hultin PM, Giorgi JV (1993) CD20 (pan-B cell) antigen is expressed at a low level on a subpopulation of human T lymphocytes. Cytometry 14:196–204
Heiligenhaus A, Miserocchi E, Heinz C, Gerloni V, Kotaniemi K (2011) Treatment of severe uveitis associated with juvenile idiopathic arthritis with anti-CD20 monoclonal antibody (rituximab). Rheumatology (Oxford) 50:1390–1394
Miserocchi E, Pontikaki I, Modorati G, Bandello F, Meroni PL, Gerloni V (2011) Rituximab for uveitis. Ophthalmology 118:223–224
Miserocchi E, Modorati G, Berchicci L, Pontikaki I, Meroni P, Gerloni V (2016) Long-term treatment with rituximab in severe juvenile idiopathic arthritis-associated uveitis. Br J Ophthalmol 100:782–786
Pelegrin L, Jakob E, Schmidt-Bacher A, Schwenger V, Becker M, Max R, Lorenz HM, Mackensen F (2014) Experiences with rituximab for the treatment of autoimmune diseases with ocular involvement. J Rheumatol 41:84–90
Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, Hessey EW, Shaw T, Totoritis MC, REFLEX Trial Group (2006) Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum 54:2793–2806
Kimby E (2005) Tolerability and safety of rituximab (MabThera). Cancer Treat Rev 31:456–473
Guex-Crosier Y, Raber J, Chan CC, Kriete MS, Benichou J, Pilson RS, Kerwin JA, Waldmann TA, Hakimi J, Roberge FG (1997) Humanized antibodies against the α-chain of the IL-2 receptor and against the beta-chain shared by the IL-2 and IL-15 receptors in a monkey uveitis model of autoimmune diseases. J Immunol 158:452–458
Gallagher M, Quinones K, Cervantes-Castañeda RA, Yilmaz T, Foster CS (2007) Biological response modifier therapy for refractory childhood uveitis. Br J Ophthalmol 91:1341–1344
Sen HN, Levy-Clarke G, Faia LJ, Li Z, Yeh S, Barron KS, Ryan JG, Hammel K, Nussenblatt RB (2009) High-dose daclizumab for the treatment of juvenile idiopathic arthritis-associated active anterior uveitis. Am J Ophthalmol 148:696–703.e1
Nussenblatt RB, Fortin E, Schiffman R, Rizzo L, Smith J, Van Veldhuisen P, Sran P, Yaffe A, Goldman CK, Waldmann TA, Whitcup SM (1999) Treatment of noninfectious intermediate and posterior uveitis with the humanized anti-tac mAb: a phase I/II clinical trial. Proc Natl Acad Sci U S A 96:7462–7466
Nussenblatt RB, Peterson JS, Foster CS, Rao NA, See RF, Letko E, Buggage RR (2005) Initial evaluation of subcutaneous daclizumab treatments for noninfectious uveitis: a multicenter noncomparative interventional case series. Ophthalmology 112:764–770
Fabiani C, Sota J, Tosi GM, Franceschini R, Frediani B, Galeazzi M, Rigante D, Cantarini L (2017) The emerging role of interleukin (IL)-1 in the pathogenesis and treatment of inflammatory and degenerative eye diseases. Clin Rheumatol 36:2307–2318
Brambilla A, Caputo R, Cimaz R, Simonini G (2016) Canakinumab for childhood sight-threatening refractory uveitis: a case series. J Rheumatol 43:1445–1447
Guedes MC, Borrego LM, Proença RD (2016) Roles of interleukin-17 in uveitis. Indian J Ophthalmol 64:628–634
Pepple KL, Lin P (2018) Targeting Interleukin-23 in the treatment of noninfectious uveitis. Ophthalmology 125:1977–1983
Wu SA, Yeh KW, Lee WI, Yao TC, Huang JL (2016) Persistent improper upregulation of Th17 and TReg cells in patients with juvenile idiopathic arthritis. J Microbiol Immunol Infect 49:402–408
Constantin T, Foeldvari I, Anton J, de Boer J, Czitrom-Guillaume S, Edelsten C, Gepstein R, Heiligenhaus A, Pilkington CA, Simonini G, Uziel Y, Vastert SJ, Wulffraat NM, Haasnoot AM, Walscheid K, Pálinkás A, Pattani R, Györgyi Z, Kozma R, Boom V, Ponyi A, Ravelli A, Ramanan AV (2018) Consensus-based recommendations for the management of uveitis associated with juvenile idiopathic arthritis: the SHARE initiative. Ann Rheum Dis 77:1107–1117
Ravelli A, Consolaro A, Horneff G, Laxer RM, Lovell DJ, Wulffraat NM, Akikusa JD, Al-Mayouf SM, Antón J, Avcin T, Berard RA, Beresford MW, Burgos-Vargas R, Cimaz R, De Benedetti F, Demirkaya E, Foell D, Itoh Y, Lahdenne P, Morgan EM, Quartier P, Ruperto N, Russo R, Saad-Magalhães C, Sawhney S, Scott C, Shenoi S, Swart JF, Uziel Y, Vastert SJ, Smolen JS (2018) Treating juvenile idiopathic arthritis to target: recommendations of an international task force. Ann Rheum Dis 77:819–828
Thorne JE, Woreta F, Kedhar SR, Dunn JP, Jabs DA (2007) Juvenile idiopathic arthritis-associated uveitis: incidence of ocular complications and visual acuity loss. Am J Ophthalmol 143:840–846
Levy-Clarke G, Jabs DA, Read RW, Rosenbaum JT, Vitale A, Van Gelder RN (2014) Expert panel recommendations for the use of anti-tumor necrosis factor biologic agents in patients with ocular inflammatory disorders. Ophthalmology 121:785–796
Simonini G, Druce K, Cimaz R, Macfarlane GJ, Jones GT (2014) Current evidence of anti-tumor necrosis factor α treatment efficacy in childhood chronic uveitis: a systematic review and meta-analysis approach of individual drugs. Arthritis Care Res 66:1073–1084
Ramanan AV, Dick AD, Jones AP, McKay A, Williamson PR, Compeyrot-Lacassagne S, Hardwick B, Hickey H, Hughes D, Woo P, Benton D, Edelsten C, Beresford MW, SYCAMORE Study Group (2017) Adalimumab plus methotrexate for uveitis in juvenile idiopathic arthritis. N Engl J Med 376:1637–1646
Leinonen ST, Aalto K, Kotaniemi KM, Kivelä TT (2017) Anti-adalimumab antibodies in juvenile idiopathic arthritis-related uveitis. Clin Exp Rheumatol 35:1043–1046
Skrabl-Baumgartner A, Erwa W, Muntean W, Jahnel J (2015) Anti-adalimumab antibodies in juvenile idiopathic arthritis: frequent association with loss of response. Scand J Rheumatol 44:359–362
Correll CK, Bullock DR, Cafferty RM, Vehe RK (2018) Safety of weekly adalimumab in the treatment of juvenile idiopathic arthritis and pediatric chronic uveitis. Clin Rheumatol 37:549–553
Dhingra N, Morgan J, Dick AD (2009) Switching biologic agents for uveitis. Eye 23:1868–1870
Miserocchi E, Modorati G, Pontikaki I, Meroni PL, Gerloni V (2014) Long-term treatment with golimumab for severe uveitis. Ocul Immunol Inflamm 22:90–95
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gaggiano, C., Rigante, D., Tosi, G.M. et al. Treating juvenile idiopathic arthritis (JIA)-related uveitis beyond TNF-α inhibition: a narrative review. Clin Rheumatol 39, 327–337 (2020). https://doi.org/10.1007/s10067-019-04763-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-019-04763-3