
Abstract
Objectives
The frequency of different vasculitides and their characteristics vary among different regions. The identification of geographic disparities of disease phenotypes helps the development of international criteria, allowing the classification of patients of different ethnicities. This study aimed to describe the frequency, characteristics, course, response to treatment, and outcome of the different adulthood vasculitides in Egypt.
Methods
This was a multicenter study in which the medical records of adult Egyptian patients diagnosed with vasculitis between 2002 and 2018 were retrospectively reviewed.
Results
The most frequent vasculitides in Egypt were Behçet’s disease (76%), hepatitis C virus vasculitis (13.9%), and granulomatosis with polyangiitis (3.9%). Most patients (73.8%) had a major event at the time of diagnosis. Generalized granulomatosis with polyangiitis was more common than the localized type (90% versus 10%, respectively). The aortic arch and its branches were the most common affected sites of Takayasu arteritis. Of vasculitides, Behçet’s disease and giant cell arteritis were associated with the greatest rates of relapse (62.7% and 33.3%, respectively). Delayed diagnosis and permanent organ damage were reported in 69.9% and 68.9% of patients, respectively. A low mortality rate was noted (1.3%).
Conclusions
The most common types of adulthood vasculitides in Egypt are Behçet’s disease, hepatitis C virus vasculitis, and granulomatosis with polyangiitis. Major organ involvement is frequent. Delayed diagnosis and permanent organ damage are common.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vasculitides are heterogeneous disorders characterized by the destructive inflammation of blood vessels. There are primary and secondary types: secondary vasculitides have probable etiologies, including infections, medications, collagen diseases, and malignancies, while primary types have unknown etiologies [1]. The frequency and characteristics of vasculitides show regional variations [2]. Most of the data about vasculitides are derived from Europeans [3]. The identification of geographic disparities of a disease helps the development of international classification criteria [4]. This study aimed to describe the frequency, characteristics, treatment, course, and outcome of the different vasculitides encountered among adults in Egypt.
Materials and methods
Study design and data acquisition
This was a retrospective cohort study. We retrospectively reviewed the medical records of adult Egyptian patients diagnosed with vasculitis between 2002 and 2018; the study included cases of primary vasculitis and cases of infection-induced vasculitis. Data were collected from three hospitals: Kasr Al Ainy Hospital, Cairo University; Tanta University Hospital; and Assuit University Hospital. Vasculitides were classified using the 1990 American College of Rheumatology criteria [5,6,7,8,9] and the 2008 International Criteria for the Classification of Behçet’s Disease (BD) [10]. Microscopic polyangiitis (MPA) was defined according to the 2012 International Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitides [1]. Epidemiological, clinical, laboratory, and imaging characteristics were recorded.
Disease onset was defined as the time of the development of the first disease manifestation. Delayed diagnosis was considered if a period of ≥ 3 months had elapsed between the disease onset and diagnosis. Disease duration was defined as the duration from the disease onset to the last visit/death.
The affection of a system included its parenchymal and vascular involvement. Major organ involvement referred to life- or organ-threatening organ involvement. The duration from disease onset to the development of the first major event was recorded.
Patients were treated according to the recommendations at the time of presentation. Low-dose corticosteroid (CS) treatment was defined as oral prednisolone ≤ 7.5 mg/day (mg/d); moderate dose, > 7.5–30 mg/d; high dose, > 30–100 mg/d; and very high dose, > 100 mg/d. Intravenous (IV) CS pulse treatment referred to an IV infusion of 250–1000 mg of methylprednisolone/day [11].
Remission was defined as the resolution of signs of inflammation that should have persisted for ≥ 3 months [12]. Relapse referred to the development of a new manifestation requiring systemic therapy adjustment. Organ damage referred to a permanent disease- or treatment-induced organ abnormality.
The study was approved by the local Research Ethics Committee (N-30-2016), and it conformed to the provisions of the Declaration of Helsinki.
Statistical analysis
Categorical variables are described in terms of frequencies and percentages. Numerical variables are described in terms of the median and interquartile range (IQR) due to skewed data distributions. Comparisons were performed using the chi-square test and calculated odds ratios and 95% confidence intervals. A two-tailed probability value (p value) was used, and p values < 0.05 were considered statistically significant. Binary logistic regression analysis was performed to eliminate the effect of confounders. All statistical calculations were performed using SPSS (Statistical Package for the Social Science; SPSS, Inc., Chicago, IL, USA) version 15 for Microsoft Windows.
Results
This study included 309 patients. Figure 1 shows the frequency of the different vasculitides. Demographic data are shown in Table 1. Delayed diagnosis was reported in 216 (69.9%) patients: 146 (67.6%) rural patients and 70 (32.4%) urban patients. The median duration from the first disease manifestation to diagnosis was 24 months (IQR, 9–48). The most common cause of delayed diagnosis was an inexperienced physician, which was noted in 190 (88%) patients with delayed diagnosis. Of the 309 patients, 227 (73.5%) developed a major event before diagnosis; a median duration of 5 months (IQR, 1–24) elapsed between disease onset and the development of a major event. The median follow-up duration was 21 months (IQR, 5–48).

Frequency of the different vasculitides. BD, Behçet’s disease; HCV, hepatitis C virus; GPA, granulomatosis with polyangiitis; TA, Takayasu arteritis; EGPA, eosinophilic granulomatosis with polyangiitis; GCA, giant cell arteritis; HSV, hypersensitivity vasculitis; PAN, polyarteritis nodosa; MPA, microscopic polyangiitis
The cumulative clinical manifestations are shown in Table 2. Table 3 shows the first induction therapies and the response to treatment in active patients.
Cumulative organ damage developed in 213 (68.9%) patients. Table 4 shows the frequency of damage of the different systems for the different vasculitides.
Among 226 patients who were followed regularly until the time of data collection, we compared patients with and without organ damage regarding the demographic, clinical, and treatment-related variables (Table 5). Although several parameters were associated with damage accrual, binary logistic regression analysis revealed the development of peripheral arterial disease (PAD) (p = 0.022), neurological disease (p = 0.046), and a major event after the achievement of remission (p = 0.015) as the only variables associated with the occurrence of permanent organ damage.
Concerning mortality, seven deaths were reported during the follow-up period: four BD patients, one patient with granulomatosis with polyangiitis (GPA), one patient with Takayasu arteritis (TA), and two cases of hepatitis C virus (HCV) vasculitis. Concerning the BD patients, one patient died following surgical resection due to intestinal perforation; another patient had neurological involvement; the third patient died following the therapeutic embolization of a pulmonary mycetoma; and the last patient died following the surgical repair of an aortic aneurysm. The GPA patient died secondary to alveolar hemorrhage. The TA patient died due to leukemia. One patient with HCV vasculitis had renal impairment, and the other patient had limb gangrene.
Discussion
Different vasculitides show geographic disparities regarding the frequency and disease phenotype. Scarce data are available for the Arab population [13,14,15]. Our study aimed to describe the frequency, characteristics, course, response to treatment, and outcome of the different adulthood vasculitides found in Egypt. We found that the most frequent vasculitides in Egypt are BD (76%), HCV vasculitis (13.9%), and GPA (3.9%); other types of vasculitides are uncommon.
Behçet’s disease
BD was the most common vasculitis in our cohort (76.1%). Our results are in agreement with those reported in the literature [16]. The highest frequency of BD was observed along the Silk Road, especially in Turkey (421/100,000), followed by Iran, where BD patients constitute 63% of primary vasculitis patients with a prevalence of 8/10,000. Moreover, BD is very frequent in Saudi Arabia, Iraq, and Japan [17]. In Japan, BD is one of the most common causes of uveitis, with a prevalence of 11.9/100,000 [18]. In an Indian study, BD represented approximately 13.62% of primary vasculitis patients [19]. In contrast to our findings, BD is rare among Australian Aboriginals and black Africans [17].
In this study, BD patients had a young age of onset (median, 26 years; IQR, 22–32). This finding is in accordance with that of an Iranian study, in which the mean (± standard deviation [SD]) age of onset was 26 ± 11.3 years [20]. However, a late age of onset was observed in an Indian study [19].
Our cohort of BD patients had a male predominance (male to female ratio, 5.5:1). This result coincides with those of several reports that have documented a male predominance in the Middle East. In contrast, women are more frequently affected in the Far East [21]; no predominance in terms of sex has been found in Iran [20] or India [19].
Orogenital ulcers were almost universal in our patients (99.6%); ocular involvement (59.6%), musculoskeletal affection (40.4%), deep venous thrombosis (28.9%), and neurological involvement (23.4%) were also common.
Caucasians more frequently exhibit genital ulcers [22], venous thrombosis, arterial affection [23], and neurological involvement [24]. Cardiac involvement is more frequent in Mediterranean and Asian populations [4]. In India, BD is mainly a mucocutaneous disease. Furthermore, pathergy test positivity showed racial variation; positive test results were detected in 37.5% of our patients. This result is in accordance with the literature; the highest frequency of positive pathergy test results was reported along the Silk Road, with a prevalence exceeding 50%, while the lowest rates of positive results have been reported in American, Northern European, Australian, and Indian populations [19].
Major organ involvement was common in our patients on presentation (74%) and during follow-up (36.2%). These results are in agreement with the observation that a high disease severity is observed along the Silk Road [3].
HCV vasculitis
HCV vasculitis represented the second most common type of vasculitis in our cohort. In Europe, HCV cryoglobulinemia is more common among Southern Europeans [25]. In our cohort, the female to male ratio was 1.9:1; a female predominance has been reported by some authors [26], while no difference between the sexes was found by others [27]. The most common manifestations in our cohort were cutaneous, followed by musculoskeletal, neurological, and peripheral arterial manifestations. The characteristics in our cohort are in agreement with those previously reported by others, with the exception of a lower frequency of renal affection in this study. Skin involvement was the disease hallmark (95% of patients); nephritis was reported in 35–60% of patients; and peripheral neuropathy was common [28]. In an Egyptian study of HCV cryoglobulinemic vasculitis, all patients had skin lesions, arthritis, and peripheral neuropathy; nephritis developed in 66.6% of patients [26]. Although major organ involvement was found in 65.1% of patients in the current study, only two cases of mortality were found. These results are in contrast to those reported by Terrier and his colleagues, in which serious organ involvement was less common (10%); however, they found a higher mortality rate, ranging between 20 and 80% [29].
Antineutrophil cytoplasmic antibody–associated vasculitis
The most prevalent type of antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV) in our cohort was GPA, followed by eosinophilic granulomatosis with polyangiitis (EGPA) and MPA; GPA represented the third most common type of vasculitis in our cohort.
The incidence of AAV is higher in Europe and the United States of America (USA); however, it is rare among Africans. Furthermore, GPA is more frequent among Caucasians, while MPA is more common among non-Caucasians [2]. However, GPA is considered the most frequent AAV in India [19], while the least frequent type is EGPA [30]. A latitude gradient has been described, with a higher prevalence of MPA noted in Southern than Northern Europe and the reverse being true for GPA [31].
In the current study, the median (IQR) ages of onset of GPA and EGPA were younger than those reported in other countries, at 29.5 (22.5–44.5) years and 35 (27–60) years for GPA and EGPA, respectively. The mean age of onset of GPA was reported to be 31.5 years (SD, 41.5) in India [32] and 46.3 years (SD, 16.8) in Greece [12], and it ranged between 43 and 58.7 years among Hispanics. The mean age of onset of MPA among Latinos has been reported to be 60 years (SD, 11.94) [30].
A slight female predominance was found among our GPA patients; the female to male ratio was 1.4:1. A higher female to male ratio has been reported in India (14:1) [32]; in contrast, a low female to male ratio was found in Greece (16:21) [12] and among Hispanics (29:36 and 21:35 in 2 different studies). Among Latinos, MPA has a female predominance, with a female to male ratio of 44:16 [30].
Regarding disease severity, AAV tends to have a higher disease activity and more severe organ involvement in Hispanics [30]. In our GPA patients, systemic disease was more common than localized disease, at a ratio of 11:1. In contrast, it has been shown that limited GPA is more prevalent than systemic GPA in Japan.
Regarding our GPA patients, the most common manifestations were mucocutaneous (75%), musculoskeletal (66.7%), renal (66.7%), upper respiratory tract (URT) (58.3%), pulmonary (58.3%), and constitutional (58.3%). In India, the most frequent manifestations were constitutional (64–89%), pulmonary (84–94%), URT (55–88%), and renal (65–73%) [32]. In a study of Hispanic patients, the main affected organs were those of the pulmonary, renal, URT, cutaneous, and nervous systems; the first three organ systems were affected in all patients, while the others were affected in 30% of patients. Ocular involvement has been reported in 48% of patients [30]; this percentage is threefold the value observed in the current cohort. In a Greek study, the most common manifestations were constitutional (92%), pulmonary (100%), URT (76%), and renal (75%) [12]. Renal involvement has been reported to be more common among European patients (77%) than Japanese patients (12–63%) [33]; in contrast, Japanese patients were older at diagnosis and more frequently showed pulmonary involvement than patients in the United Kingdom (UK) [34].
In our GPA patients, 91.7% and 57.1% of patients developed a major event on presentation and during follow-up, respectively. Most major events affected the pulmonary and neurological systems. In a Greek study, 44% of patients developed serious events during a follow-up period of 42 months; the most common event was renal failure (37.5%) [12].
In our GPA patients, IV cyclophosphamide (CYC) was the mainstay of therapy; the response rate to the first induction therapy was 72.7%. In a Greek study, oral and IV CYC were equally used, and out of 35 patients completing the induction therapy, 29 (82.8%) patients achieved remission. The authors concluded that the characteristics and outcome of GPA in Greece are similar to those in other populations, except for a higher frequency of fever among Greek patients [12]. In a study from Latin America, IV CS and CYC were the main forms of treatment [30].
In a review of vasculitis in Hispanic patients, the main affected organs in patients with MPA were the kidneys (100%), peripheral nervous system (PNS) (61.1%), lungs (50%), and skin (44.4) in one included study; in another included study, the main affected organs were the PNS (86.7%), musculoskeletal system (65%), kidneys (56.7%), skin (41.7%), and lungs (38.3%). Pulmonary renal syndrome developed in 16.7% and 16% of patients in the other two included studies. Over a mean duration of 47.46 months (SD, 33.91), 43.3% of patients relapsed [30].
Concerning pulmonary involvement in AAV, pulmonary fibrosis is more prevalent in Asians, while alveolar hemorrhage is more frequent among patients from the UK [35]. In two studies comparing patients from the UK and Japan with MPA [31] and GPA [34], those from the UK had more severe renal affection; greater use of immunosuppressants, biological agents, and plasmapheresis; and greater cumulative CYC doses. While ocular affection was more common among the MPA patients from the UK, it was more frequent among Japanese GPA patients. Regarding GPA, relapse was more frequent but the survival rate was higher among UK patients. Concerning MPA, the rates of renal and patient survival were the same among Japanese and European patients despite less aggressive therapy being administered in Japan.
Concerning GPA, cytoplasmic/c-ANCA was detected in 90% of patients in the current study. It has been reported that perinuclear/p-ANCA and anti-myeloperoxidase/MPO antibodies are more prevalent in the Far East than in the UK, while the reverse has been noted for c-ANCA and anti-proteinase 3/anti-PR3 antibodies [36].
The mortality rate of GPA patients was lower in our study (8.3%) than in other reports. Death was reported in 18% of Hispanic patients during a follow-up period of 15 months. In another study of Hispanic patients, the reported survival rates were 90% for 5 years, 84% for 10 years, and 63% for 20 years [31]. A very high mortality rate (50%) was reported for Indian patients [32]. For Greek patients, 10% of patients died during a median follow-up period of 42 months; 75% of the deaths were due to disease activity [12].
In a study of AAV from Latin America, death was reported in 17% of patients during a follow-up period of 20 months; pulmonary renal syndrome was the main cause of death [30].
Takayasu arteritis
TA was an uncommon type of vasculitis in our cohort (1.3%). However, it is considered the most common vasculitis in Japan, Korea, China, India, and Latin America, while it is uncommon in Europe and the USA. The incidence of TA in Japan is 150/million [37]. In India, TA represents approximately 20% of primary vasculitis cases and is considered the most common cause of renovascular hypertension [19]. In Africa, the highest prevalence is observed among South Africans [30].
In this study, TA was more frequent among females (85.7%), with a median (IQR) age of onset of 35 (24–40) years. These results are in agreement with those of previous reports. TA is considered a typical disease of young women, with a median age of onset of 25 years [37]. A striking female to male ratio of 9:1 has been reported in Japan. In Iran, the ratio is slightly lower (7.6:1), while the reverse has been found in India (1:1.3) [38]. Among Latinos, the female to male ratio ranges between 2.8:1 and 24:1 [30]. In India and China, males have an older age of onset [39].
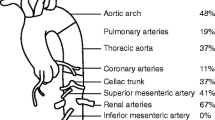
In this study, the aortic arch and its branches were the most common affected sites of the disease, which is similar to previous reports from Japan, the USA, North Europe [39], and Latin America [30]. In contrast, the abdominal aorta is the most common site affected in India [39].
In the current study, the most common manifestations were peripheral arterial, constitutional, cardiac, neurological, and aortic manifestations. The most commonly used drugs for induction were IV CYC and methotrexate (MTX), and all patients responded well to treatment. One patient underwent bilateral lower limb amputation, and one underwent unilateral upper limb amputation. Despite the high morbidity rate in our cohort, only one case of mortality was reported during the follow-up period and was not directly related to disease activity.
In Latin Americans, the clinical spectrum of disease included constitutional manifestations, peripheral arterial disease, heart disease, and systemic hypertension in 15%, 32%, 9%, and 6% of patients, respectively, in one study and in 27.5%, 85%, 35.5 and 29% of patients, respectively, in another study. The main treatments were CS, being administered to 70% and 97.05% of patients in the two studies, and MTX, being administered to 26.47% and 40% of patients in the two studies. Surgery was performed in 26.47% and 33.3% of patients in the two studies. Delayed diagnosis of up to 10 years was reported, and 58% of all patients responded well to treatment [30]. Among Japanese patients, the 15-year survival rate is reportedly 83% [39], while that among North Americans is reportedly 85% [40].
Giant cell arteritis
In this study, three female patients had giant cell arteritis (GCA); the median age of onset was 66 years (IQR, 59–68). GCA is considered the most frequent primary vasculitis in Europe, the USA, and Canada. In contrast, GCA is rare in Asia, Africa, and Latin America. A latitude gradient has been described, with the highest incidence reported in the Northern Hemisphere, especially among Scandinavians (15–35/100,000) [2].
A slight female predominance is suggested, with a female to male ratio of 2:1; however, sex differences could not be found among Asians. The mean age of onset has been reported to be 72 years; however, this value was reported to be slightly lower (63.97 years) in Asian populations [41].
Polyarteritis nodosa
Our cohort included one case of hepatitis B virus (HBV)–negative polyarteritis nodosa (PAN) with an age of onset of 72 years; the patient had constitutional, cutaneous, musculoskeletal, neurological, and renal manifestations. PAN is more frequent among Europeans than other ethnicities; the incidence of PAN in Europe and the USA is 2–9/million [42]. A much higher incidence of 77/million has been described in Alaska, where HBV infection is endemic [43]. In an Indian study, systemic and cutaneous PAN represented 8.83% and 1.22% of primary vasculitides, respectively [19].
In a Swedish study, the range of age of onset was 40–60 years, with no sex predilection [42]. In a Greek study, 22% of PAN patients had HBV infection [12]. In a Latin American study including 20 patients, the female to male ratio was 3:2; 16 patients had systemic PAN, and four had cutaneous PAN. Four of the patients with systemic PAN died due to renal and cardiac involvement and septicemia. Eight patients were tested for HBV; two had positive results [30]. In an Indian study, the mean age of onset was lower than that in other populations (33.75 years); the female to male ratio was reversed (1:4.5); and the most common disease manifestations were constitutional manifestations (60–100%), hypertension (60–100%), mononeuritis multiplex (60–100%), and cutaneous manifestations (41–75%) [44].
Although the aim of our study was to present the characteristics of systemic vasculitides in adults, it is noteworthy that Kawasaki disease, a unique type of systemic vasculitides in the pediatric population, was reported in the Egyptian children in the form of case series with the predominance of the atypical form of the disease [45].
The authors can conclude that the most frequent vasculitides in the current population were BD, HCV vasculitis, and GPA. Delayed diagnosis, the development of major events at the time of diagnosis, and the occurrence of permanent organ damage were common; however, the mortality rate was low. Early diagnosis, the institution of immunosuppressants, and close follow-up are necessary to prevent major organ affection and damage.
The limitations of this study include its retrospective nature and the relatively small sample size. Other multicenter prospective studies involving a larger population size are recommended to verify these results.
References
Jennette JC, Falk RJ, Bacon PA (2013) 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 65(1):1–11
Kobayashi S, Fujimoto S (2013) Epidemiology of vasculitides: differences between Japan, Europe and North America. Clin Exp Nephrol 17:611–614
Watts RA (2005) Epidemiology of vasculitis in India-what is known? J Indian Rheumatol Assoc 13:8–15
Leonardo NM, McNeil J (2015) Behçet’s disease: is there geographical variation? a review far from the Silk Road. Int J Rheumatol 2015:945262. https://doi.org/10.1155/2015/945262
Arend WP, Michel BA, Bloch DA (1990) American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum 33(8):1129–1134
Lightfoot RW, Michel BA, Bloch DA, Hunder GG, Zvaifler NJ, McShane DJ et al (1990) The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum 33:1088–1093
Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, Calabrese LH, Fries JF, Lie JT, Lightfoot RW Jr (1990) The American College of Rheumatology 1990 criteria for the classification of wegener's granulomatosis. Arthritis Rheum 33(8):1101–1107
Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, Calabrese LH, Edworthy SM, Fauci AS, Leavitt RY (1990) The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 33(8):1094–1100
Calabrese LH, Michel BA, Bloch DA, Arend WP, Edworthy SM, Fauci AS, Fries JF, Hunder GG, Leavitt RY, Lie JT (1990) American College of Rheumatology 1990 criteria for the classification of hypersensitivity vasculitis. Arthritis Rheum 33:1108–1113
International Team for the Revision of International Criteria for Behçet’s Disease (2008) Clinical manifestations of Behçet’s disease. The ITR-ICBD report. Clin Exp Rheumatol 26(Suppl 50):S1–S18
Buttgereit F, da Silva JA, Boers M, Burmester GR, Cutolo M, Jacobs J et al (2002) Standardized nomenclature for glucocorticoid treatment regimens: current questions and tentative answers in rheumatology. Ann Rheum Dis 61(8):718–722
Boki KA, Dafni U, Karpouzas GA, Papasteriades C, Papasteriades C, Drosos AA et al (1997) Necrotizing vasculitis in Greece: clinical, immunological and immunogenetic aspects. A study of 66 patients. Br J Rheumatol 36(10):1059–1066
Chaudhry IA, Shamsi FA, Elzaridi E, Arat YO, Bosley TM, Riley FC (2007) Epidemiology of giant-cell arteritis in an Arab population: a 22-year study. Br J Ophthalmol 91:715–718
Khalifa M, Karmani M, Jaafoura NG, Kaabia N, Letaief AO, Bahri F; Study Group of GCA in Tunisia (2009) Epidemiological and clinical features of giant cell arteritis in Tunisia. Eur J Intern Med 20:208–212
Shahin AA, Zayed HS, Elrefai RM, Taher H, Elsaie A, Senara SH, Fathi HM, Omar G, Abd Elazeem MI (2018) The distribution and outcome of vasculitic syndromes among Egyptians: a multi-centre study including 630 patients. Egyptian Rheumatol 40(4):243–248
Hatemi G, Seyahi E, Fresko I, Talarico R, Hamuryudan V (2015) Behçet's syndrome: a critical digest of the 2014–2015 literature. Clin Exp Rheumatol 33:S3–S14
Mohammad A, Mandl T, Sturfelt G, Segelmark M (2013) Incidence, prevalence and clinical characteristics of Behçet’s disease in southern Sweden. Rheumatology (Oxford) 52(2):304–310
Kitaichi N, Miyazaki A, Iwata D, Ohno S, Stanford MR, Chams H et al (2007) Ocular features of Behçet’s disease: an international collaborative study. Br J Ophthalmol 91(12):1579–1582
Joshi VR, Mittal G (2006) Vasculitis—Indian perspective. J Assoc Physicians India 54(Suppl):12–14
Davatchi F, Shahram F, Chams-Davatchi C, Shams H, Nadji A, Akhlaghi M et al (2010) Behçet’s disease in Iran: analysis of 6500 cases. Int J Rheum Dis 13:367–373
Bang D, Oh S, Lee KH, Lee ES, Lee S (2003) Influence of sex on patients with Behçet's disease in Kore. J Korean Med Sci 18(2):231–235
Davatchi F, Shahram F, Chams-Davatchi C, Shams H, Nadji A, Akhlaghi M, Faezi T, Ghodsi Z, Faridar A, Ashofteh F, Sadeghi Abdollahi B (2010) Behçet’s disease: from east to west. Clin Rheumatol 29(8):823–833
Ames PRJ, Steuer A, Pap A, Denman AM (2001) Thrombosis in Behçet’s disease: a retrospective survey from a single UK centre. Rheumatology 40(6):652–655
Joseph FG, Scolding NJ (2007) Neuro-Behçet’s disease in Caucasians: a study of 22 patients. Eur J Neurol 14(2):174–180
Sansonno D, Dammacco F (2005) Hepatitis C virus, cryoglobulinemia, and vasculitis: immune complex relations. Lancet Infect Dis 5:227–236
Mohammed RH, ElMakhzangy HI, Gamal A, Mekky F, El Kassas M, Mohammed N et al (2010) Prevalence of rheumatologic manifestations of chronic hepatitis C virus infection among Egyptians. Clin Rheumatol 29(12):1373–1380
Terrier B, Jehan F, Munteanu M, Geri G, Saadoun D, Sène D et al (2012) Low 25-hydroxyvitamin D serum levels correlate with the presence of extrahepatic manifestations in chronic hepatitis C virus infection. Rheumatology (Oxford) 51(11):2083–2090
Johnson R, Gretch D, Yamabe H, Hart J, Bacchi CE, Hartwell P et al (1993) Membranoproliferative glomerulonephritis associated with hepatitis C virus infection. N Engl J Med 328:465–470
Terrier B, Semoun O, Saadoun D, Font J (2011) Prognostic factors in patients with hepatitis C virus infection and systemic vasculitis. Arthritis Rheum 63(6):1748–1757
Gamarra AI, Coral P, Quintana G, Toro CE, Flores LF, Matteson EL et al (2010) History of primary vasculitis in Latin America. Med Sci Monit 16(3):RA58–RA72
Furuta S, Chaudhry AN, Hamano Y, Fujimoto S, Nagafuchi H, Makino H, Matsuo S, Ozaki S, Endo T, Muso E, Ito C, Kusano E, Yamagata M, Ikeda K, Kashiwakuma D, Iwamoto I, Westman K, Jayne D (2014) Comparison of phenotype and outcome in microscopic polyangiitis between Europe and Japan. J Rheumatol 41:325–333
Handa R, Wali JP, Biswas A, Aggarwal P, Wig N (1997) Wegener’s granulomatosis- a clinicopathologic study. J Assoc Physicians India 45:536–539
Ishida Y, Katada A, Kishibe K, Imada M, Hayashi T, Nonaka S, Harabuchi Y (2004) Wegener’s granulomatosis with otolaryngological symptoms. Practica Oto-Rhino-Laryngol 97:997–1005
Furuta S, Chaudhry AN, Arimura Y, Dobashi H, Fujimoto S, Homma S, Rasmussen N, Jayne DR (2017) Comparison of the phenotype and outcome of granulomatosis with polyangiitis between UK and Japanese cohorts. J Rheumatol 44:216–222
Arulkumaran N, Periselneris N, Gaskin G, Strickland N, Ind PW, Pusey CD, Salama AD (2011) Interstitial lung disease and ANCA-associated vasculitis: a retrospective observational cohort study. Rheumatology (Oxford) 50:2035–2043
Oh JS, Lee CK, Kim YG, Nah SS, Moon HB, Yoo B (2009) Clinical features and outcomes of microscopic polyangiitis in Korea. J Korean Med Sci 24:269–274
Kerr GS, Hallahan CW, Giordano J, Leavitt RY, Fauci AS, Rottem M, Hoffman GS (1994) Takayasu arteritis. Ann Intern Med 120:919–929
Numano F (2002) The story of Takayasu arteritis. Rheumatology (Oxford) 41:103–106
Numano F (1997) Differences in clinical presentation and outcome in different countries for Takayasu’s arteritis. Curr Opin Rheumatol 9:12–15
Ogino H, Matsuda H, Minatoya K, Sasaki H, Tanaka H, Matsumura Y, Ishibashi-Ueda H, Kobayashi J, Yagihara T, Kitamura S (2008) Overview of late outcome of medical and surgical treatment for Takayasu arteritis. Circ 118:2738–2747
Hellmann DB (2017) Giant cell arteritis, polymyalgia rheumatica, and Takayasu’s arteritis. In: Firestein GS, Budd RC, Gabriel SE, Mcinnes LB, O'Dell JR et al (eds) Kelley & Firestein’s textbook of rheumatology, 10th edn. Elsevier, Philadelphia, pp 1520–1540
Mohammad AJ, Jacobsson LT, Mahr AD, Sturfelt G, Segelmark M (2007) Prevalence of Wegener’s granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology (Oxford) 46:1329–1337
McMahon BJ, Heyward WL, Templin DW, Clement D, Lanier AP (1989) Hepatitis B-associated polyarteritis nodosa in Alaskan eskimos: clinical and epidemiologic features and long-term follow-up. Hepatology 9:97–101
Handa R, Wali JP, Gupta SD, Dinda AK, Aggarwal P, Wig N, Biswas A (2001) Classic polyarteritis nodosa and microscopic polyangiitis-a clinicopathologic study. J Assoc Physicians India 49:314–319
Attia TH, Morsy SM, Hassan BA, Ali ASA (2018) Kawasaki disease among Egyptian children: a case series. Glob Cardiol Sci Pract 2017, 25. https://doi.org/10.21542/gcsp.2017.25
Acknowledgements
The authors would like to thank Prof Dr. Geilan Mahmoud Abd El-Moniem, Professor of Rheumatology and Clinical Immunology, Cairo University, for reviewing the work and providing valuable advice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Attia, D.H.S., Abdel Noor, R.A. & Salah, S. Shedding light on vasculitis in Egypt: a multicenter retrospective cohort study of characteristics, management, and outcome. Clin Rheumatol 38, 1675–1684 (2019). https://doi.org/10.1007/s10067-019-04441-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-019-04441-4