
Abstract
To investigate the clinical features, risk factors and outcomes of patients with interstitial pneumonia with autoimmune features (IPAF). A total of 1429 patients with idiopathic interstitial pneumonia (IIP) and undifferentiated connective tissue disease-associated interstitial lung disease (UCTD-ILD) were screened to identify patients who met IPAF criteria. Clinical, serological, and morphological features of patients with IPAF were characterized. Outcomes between patients with IPAF, UCTD-ILD, and IIP who were divided into idiopathic pulmonary fibrosis (IPF) and non-IPF groups were compared using survival as an endpoint. Patients with IPAF were much common in young female and had lower percentage of ever smoking and a significantly shorter survival than those with non-IPAF (P < 0.001). Subgroup analysis revealed that IPAF cohort survival was worse than that in non-IPF (P < 0.001), but better than that in IPF (P < 0.001). In IPAF cohort, the most common systemic symptom and serological abnormality were Raynaud’s phenomenon (12.9%) and ANA ≥ 1:320 (49.2%); the most frequent high-resolution computed tomography (HRCT) pattern was nonspecific interstitial pneumonia (NSIP) (61.6%). Multivariate analysis indicated that several factors including age, smoking history, organizing pneumonia (OP) pattern in HRCT, and anti-RNP positivity were independently associated with significantly worse survival. IPAF had the distinct clinical features and outcomes compared with other groups of ILD. Additional studies should be needed to explore the underlying autoimmune mechanism and to determine risk stratification in future clinical research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Interstitial pneumonia with autoimmune features (IPAF) is a new term that was proposed by the joint of European Respiratory Society and American Thoracic Society (ERS/ATS) research statement. It is based on a combination of clinical, serological, and morphological domains that depict patients who have interstitial lung disease (ILD) with autoimmune features, but not meet the specific criteria for connective tissue disease (CTD) [1]. Indeed, a variety of terms such as autoimmune-featured interstitial lung disease (AI-ILD) [2], undifferentiated connective tissue disease-associated interstitial lung disease (UCTD-ILD) [3], and lung-dominant CTD (LD-CTD) [4] had been used in studies to describe this group of patients. These terms had different but some overlapping criteria. AI-ILD was defined as patients who did not meet the American College of Rheumatology (ACR) criteria for a CTD having at least one sign or symptom suggestive of a CTD and at least one serologic test reflective of an autoimmune process [2]. UCTD-ILD was defined as having one of the specific autoantibodies (SSA, SSB, anti-Scl-70, anti-centromere, anti-RNP, or Jo-1) or a positive ENA or ANA titer, as well as symptoms or signs of CTD [3]. LD-CTD was diagnosed when any one of defined serologic autoantibodies (ANA>1:320, nucleolar-ANA, rheumatoid factor>60IU/ml, CCP, anti-Scl-70, SSA, SSB, dsDNA, anti-Smith, anti-RNP, Jo-1 and anti-centromere antibodies) at the initial evaluation was present and a specific CTD was absent [4].
As for IPAF, a consensus on the terminology and diagnostic criteria provided a solid basis for the development of uniform clinical research. Although a previous study reported the characterization of patients with IPAF [5,6,7,8], several controversies still remain. For example, whether the presence of usual interstitial pneumonia (UIP) should be included in the morphological domain [9]? Among the distinct clinical, serological, and morphological parameters, which are independently associated with the survival? Furthermore, there is no study to report the prevalence, clinical features, and outcomes in Chinese patients with IPAF. Hence, there is an urgent need to present data from Chinese populations regarding the new IPAF classification.
In the current study, we applied the diagnostic criteria of IPAF [1] to patients who were referred to the Department of Respiratory Medicine experienced in ILD in our center from September 2010 to December 2016 and who had been initially diagnosed as ILD including idiopathic ILD that was also called idiopathic interstitial pneumonia (IIP), and UCTD-ILD. We described the clinical, serological, and morphological features of IPAF and compared outcomes among patients with IPAF, UCTD-ILD, and IIP who were classified as IPF and non-IPF group based on the presence of honeycombing in chest high-resolution computed tomography (HRCT). We also evaluated the prognostic value of features in each domain of the current diagnostic criteria of IPAF.
Methods
Patient cohort
Electronic medical records of patients enrolled at the Department of Respiratory Medicine, the Affiliated Drum Tower Hospital of Nanjing University Medical School, from September 2010 to December 2016 were retrospectively reviewed. All patients receiving a diagnosis of ILD—including biopsy-proven ILD, clinical diagnosis of IIP, and UCTD-ILD—were screened. This was a retrospective study and the ethics approval was acquired in accordance with the policy of the Ethics Committee of the Affiliated Drum Tower Hospital of Nanjing University Medical School.
Diagnostic criteria
The diagnosis of ILD was confirmed through a combination of clinical, physiological, HRCT findings, and lung biopsies examinations (if available) in accordance with the criteria of ATS/ERS guidelines [10, 11]. All diagnoses were made by the multidisciplinary approach in conjunction with experienced pulmonologists, chest radiologists, and pathologists, when lung biopsies were performed. The diagnostic criteria for IPAF were based on the criteria proposed by ERS/ATS research statement [1].
All patients with CTD-ILD or UCTD-ILD were confirmed by a rheumatologist. ILD with known causes such as drug or occupational exposure, and CTD were excluded. The diagnostic criteria for CTD were based on the recommendations by ACR [12,13,14,15,16]. The diagnostic criteria of UCTD were defined as patients with features of a systemic autoimmune disease who do not meet ACR classification criteria for specific CTD [3, 17,18,19].
Data collection
Clinical information included demographics, general medical history, smoking history, clinical symptoms, and serologic autoantibodies. Treatment was recorded including systemic corticosteroids and suppressive agents using. Vital status was determined using medical record reviews and telephone communications. All patients were followed up to October 2017. Survival time was recorded from the first hospital admission.
Chest HRCT evaluation
Chest HRCTs of all patients were performed in supine position at end inspiration at the time of initial diagnosis. Two dedicated chest radiologists (K.Z. and J.H.) blindly reviewed and interpreted HRCT patterns. Based on the proposed criteria of the guidelines [11], nonspecific interstitial pneumonia (NSIP) pattern was defined as basal predominant reticular abnormalities with traction bronchiectasis, frequently associated with ground-glass attenuation; organizing pneumonia (OP) pattern was defined as bilateral patchy areas of consolidation and/or ground-glass opacities with a subpleural and lower lung zone predominance or peri-bronchovascular distributions; NSIP with OP overlapping was defined as basal predominant consolidation, associated with traction bronchiectasis and reticular abnormality; and UIP pattern was defined as basal honeycombing opacities associated with reticular abnormalities and traction bronchiectasis. No lymphoid interstitial pneumonia (LIP) HRCT pattern was found in this cohort. Any disagreements between the two chest radiologists were resolved by consensus after discussion.
Histological assessment
Among patients with IIP and UCTD, those undergoing transbronchial lung biopsies (TBLB) were analyzed and the biopsy specimen slides were reviewed by a pulmonary pathologist (F.M.) with expertise in ILD. The pathologic pattern was defined according to the guideline statement [11].
Statistical analysis
Continuous variables were compared using a two-tailed Student’s t test. Categorical variables were compared using a chi-squared test, or Fisher’s exact test when needed. The Kaplan-Meier curve and two-sided log-rank test were used for univariate survival analysis. The Cox proportional hazards model was used for univariate and multivariate survival analysis to calculate the hazard ratios (HR) and corresponding 95% confidence intervals (CI). Survival time was calculated from the date of initial diagnosis to death from any cause and transplant. For a patient who lost to follow-up, survival time was censored at the last follow-up date. P values were two-sided and considered significant if less than 0.05. All statistical analyses were performed using the SPSS statistical software, version 20.0 (SPSS Inc., Chicago, IL, USA).
Results
Patients inclusion
In total, 2644 patients with ILD in the database were screened. After exclusions, 1429 patients were identified. Among the 1429 patients, there were 177 who met the IPAF criteria and 1252 patients who did not fulfilled the IPAF criteria were defined as non-IPAF cohort. Of the 177 patients, there were 41 with UCTD-ILD, and 136 with IIP including 17 with biopsy-proven cryptogenic OP, 1 biopsy-proven NSIP, 8 with IPF, and 110 clinical diagnosis of IIP (Fig. 1).

Flowchart of the patients’ selection
The non-IPAF cohort was classified into IIP (n = 1231) and UCTD-ILD groups (n = 21), and the IIP cohort was divided into IPF (n = 235) and non-IPF groups (n = 996).
Clinical features
The demographics, smoking history, and clinical and treatment information of the IPAF, IPF, and non-IPF cohorts are summarized in Table 1. In IPAF cohort, there were 78 males (44.0%) and 99 females (56.0%) with the mean age of 60.2 ± 12.8 years and 34 (19.2%) patients reported a smoking history. IPF cohort included 202 males (86.0%) and 33 females (14.0%) with the mean age of 67.6 ± 8.6 years. One hundred forty-one (60.0%) patients with IPF had a smoking history. Thus, patients with IPAF were younger, more often female and had lower percentage of ever smoker than IPF (P < 0.001). Patients with UCTD had similar age (58.6 ± 11.2 years) and sex ratio (9 males/12 females) compared to patients with IPAF (Table 2).
The characteristics of each domain for patients with IPAF are presented in Table 3. The most prevalent systemic symptoms were Raynaud’s phenomenon (12.9%), followed by inflammatory arthritis and polyarticular morning joint stiffness ≥ 60 min (4.5%). ANA ≥ 1:320 (49.2%) was the most frequent serological abnormality, followed by anti-Ro/SSA (36.1%). In the morphological domain, NSIP (61.6%) was the most frequent HRCT pattern, followed by an OP pattern (22.0%) and an overlap of NSIP and OP (11.9%), and eight patients (4.5%) demonstrated the UIP pattern.
In the IPAF cohort, there were 18 patients that underwent TBLB histological assessment. Among them, 17 (9.6%) patients presented with OP showing the presence of connective tissue in airspaces extending into alveolar ducts and 1 (0.6%) patient presented with NSIP showing diffuse alveolar wall thickening with inflammation infiltration, no honeycombing or fibroblastic foci seen. In the 17 biopsy-proven COP patients, HRCT demonstrated patchy consolidations in basal predominance in 8 patients, HRCT demonstrated patchy consolidations and ground-glass opacities in peri-bronchovascular distributions in 7 patients, and in 2 patients, HRCT demonstrated basal predominant consolidations, associated with reticular abnormalities. In the 1 biopsy-proven NSIP patient, the HRCT demonstrated reticular abnormalities and ground-glass opacities in basal predominance distributions.
Survival analysis
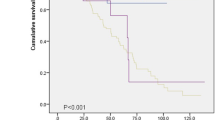
In total, 35 patients with IPAF died during the study period; the mean survival time was 295.0 weeks. In the non-IPAF cohort, 125 patients died and the mean survival time was not reached. Patients with IPAF had significantly worse survival compared to non-IPAF patients (P < 0.001, Fig. 2a). Then, we conducted the subgroup analysis of non-IPAF cohort. In the UCTD-ILD cohort, 3 patients died and the mean survival time was not reached. Patients with IPAF had similar survival to those with UCTD-ILD (P = 0.257, Fig. 2b). In the IPF cohort, 75 patients died; the mean survival time was 128.0 weeks. In non-IPF group, 50 patients died; the mean survival time was not reached. Patients with IPAF had markedly worse survival compared to non-IPF patients, but had dramatically better survival than the IPF group (P < 0.001, Fig. 2c).

Kaplan-Meier curves in different patients’ cohort. a Patients with IPAF had significantly worse survival than those with non-IPAF (P < 0.001). b Patients with IPAF had similar survival to patents with UCTD-ILD (P = 0.257). c Patients with IPAF had a significantly better survival than those with IPF, but worse than non-IPF group (P < 0.001)
Risk factors
Univariate analysis revealed that age (HR = 1.041, 95% CI 1.014–1.068, P = 0.002), smoking history (HR = 2.435, 95% CI 1.185–5.002, P = 0.015), ANA ≥ 1:320 (HR = 2.079, 95% CI 1.033–4.186, P = 0.040), anti-RNP positivity (HR = 4.140, 95% CI 1.446–11.855, P = 0.008), OP pattern in HRCT (HR = 3.712, 95% CI 1.130–12.196, P = 0.031), and the presence of pleural effusion or thickening (HR = 2.315, 95% CI 1.105–4.852, P = 0.026) were significantly associated with worse survival. After multivariable adjustment, only age (HR = 1.036, 95% CI 1.009–1.063, P = 0.007), smoking history (HR = 2.108, 95% CI 1.018–4.361, P = 0.045), anti-RNP positivity (HR = 4.737, 95% CI 1.489–12.844, P = 0.007), and OP pattern in HRCT (HR = 3.385, 95% CI 1.017–11.261, P = 0.047) remained the significant predictors for poor survival (Table 4).
Discussion
In this study, we characterized Chinese patients with IPAF from a large group that included 1429 patients diagnosed with IIP and UCTD-ILD. Approximately 12% (177/1429) of the patients fulfilled the diagnostic criteria for IPAF. Although the proportion varied among different studies [5,6,7], this study suggested that the features of CTD were not uncommon among patients with IIP and UCTD-ILD.
The current evidence indicated that patients with IPAF tended to be young and have higher female percentage and lower percentage of ever smoking than IPF. There were no significant differences of age and sex distributions between IPAF and UCTD groups. Of note, Oldham et al. showed that patients who met the IPAF criteria were older and had a higher percentage of ever smokers compared to the UCTD cohort. The inconsistent findings were mainly due to the high percentage of patients (about 30%) with IPF in their patient cohort [5]. IPF patients were common in the study by Oldham et al., mostly because their study population was drawn from a single tertiary referral center with expertise in IPF, leading IPF patients overrepresented [5]. However, in the current study, the majority of patients with IPAF were identified from patients with UCTD-ILD (41/62, 66.1%) and IIP (110/976, 11.3%), and only 8 (8/243, 3.3%) patients with IPF met the IPAF criteria.
In the current study, patients who met the IPAF criteria had a significantly worse survival than those with non-IPAF. When we did subgroup analysis, patients with IPAF had a similar survival to UCTD-ILD, a significantly better survival than IPF, but worse than non-IPF group. Our study showed that patients with IPF had the poorest survival compared with IPAF and non-IPF. Consistently, two previous studies reported that although UIP was associated with poor survival rates among patients with CTD, the prognosis in patients with the UIP pattern in CTD was still better than that of patients with IPF, suggesting that patients with IPF had the worst survival [20, 21]. Nevertheless, several recent studies reported that overall survival did not significantly differ between IPAF and IPF groups [2, 5, 7]. The possible reason for this discrepancy was that NSIP was the most frequent pattern of HRCT finding in our IPAF cohort; however, the UIP pattern had a relative high proportion in the previous study [5]. Our study supported previous findings that most patients diagnosed with “idiopathic” NSIP met the criteria of UCTD [3, 17]. Similar studies also reported that NSIP was the most frequent HRCT pattern in their IPAF cohort [6,7,8]. As the majority of the patients with IPAF in our study were NSIP (UIP only accounted 4.5%, 8/177) in HRCT, our study indicated that the underlying autoimmune process had a negative effect on the survival especially for the patients presented with non-UIP on HRCT.
In the IPAF cohort, Raynaud’s phenomenon and ANA ≥ 1:320 were the most common systemic symptom and serological abnormality, which was consistent with previous studies [5, 7]. However, they were not associated with mortality of IPAF. After multivariable adjustment, age, ever smoker, anti-RNP positivity, and OP pattern in HRCT remained significantly independent predictors for survival in the IPAF cohort. It was noteworthy that a subset of patients who presented with “idiopathic” OP pattern with or without overlapping NSIP in HRCT, who was negative of anti-JO-1 antibody, was ultimately diagnosed as polymyositis or antisynthetase syndrome [22]. Therefore, it is necessary to measure comprehensive serologic autoantibodies serially and carefully evaluate for extrapulmonary symptoms to reach a final accurate diagnosis during follow-up examinations. We hypothesized that consolidations and ground-glass opacities in OP HRCT pattern pathologically represented an active inflammatory reaction or diffuse alveolar damage caused by an underlying autoimmune process. Further radiographic-pathologic studies should be carried out to investigate the underlying autoimmune mechanism contributing to the disease pathogenesis.
The current study had several limitations. First, this was a retrospective, population-based observational study from a single center. Although clinical data was prospectively collected from the included patients, the center-specific and selection bias was inevitable (for example, interpretation of the HRCT finding and detection technology of serologic autoantibody). Future study that applies the IPAF criteria should be prospectively performed in a larger cohort from multiple institutions with diverse populations. Second, only a few subtle of patients underwent TBLB, which provided modest value in assessing the histopathological morphology. Surgery lung biopsy was not performed because of patients’ refusal or advanced age. However, all diagnoses were made in a rigorous, multidisciplinary approach including experienced clinicians and dedicated thoracic radiologists. For patients with ILD who received corticosteroid treatment, a careful follow-up was programmed to observe the response of treatment to validate the diagnosis.
In conclusion, our study suggested that IPAF had distinct clinical features and outcome compared with other groups of ILD. Patients with IPAF tended to be younger and have higher percentage of females and lower percentage of ever smoker than those with IPF. Patients with IPAF had worse survival than patients with non-IPF, but better than those with IPF. Age, smoking history, OP patterns in HRCT, and anti-RNP positivity were the independently significant prognostic factors for IPAF. Additional studies will be needed to reveal the precise mechanism of the underlying autoimmune processes, to determine prognostic model in future clinical research.
References
Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, Lee JS, Leslie KO, Lynch DA, Matteson EL, Mosca M, Noth I, Richeldi L, Strek ME, Swigris JJ, Wells AU, West SG, Collard HR, Cottin V (2015) An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 46:976–987. https://doi.org/10.1183/13993003.00150-2015
Vij R, Noth I, Strek ME (2011) Autoimmune-featured interstitial lung disease. Chest 140:1292–1299. https://doi.org/10.1378/chest.10-2662
Corte TJ, Copley SJ, Desai SR, Zappala CJ, Hansell DM, Nicholson AG, Colby TV, Renzoni E, Maher TM, Wells AU (2012) Significance of connective tissue disease features in idiopathic interstitial pneumonia. Eur Respir J 39:661–668. https://doi.org/10.1183/09031936.00174910
Omote N, Taniguchi H, Kondoh Y, Watanabe N, Sakamoto K, Kimura T, Kataoka K, Johkoh T, Fujimoto K, Fukuoka J, Otani K, Nishiyama O, Hasegawa Y (2015) Lung-dominant connective tissue disease: clinical, radiologic, and histologic features. Chest 148:1438–1446. https://doi.org/10.1378/chest.14-3174
Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, Husain AN, Montner S, Chung JH, Cottin V, Fischer A, Noth I, Vij R, Strek ME (2016) Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J 47:1767–1775. https://doi.org/10.1183/13993003.01565-2015
Chartrand S, Swigris JJ, Stanchev L, Lee JS, Brown KK, Fischer A (2016) Clinical features and natural history of interstitial pneumonia with autoimmune features: a single center experience. Respir Med 119:150–154. https://doi.org/10.1016/j.rmed.2016.09.002
Ahmad K, Barba T, Gamondes D, Ginoux M, Khouatra C, Spagnolo P, Strek M, Thivolet-Béjui F, Traclet J, Cottin V (2017) Interstitial pneumonia with autoimmune features: clinical, radiologic, and histological characteristics and outcome in a series of 57 patients. Respir Med 123:56–62. https://doi.org/10.1016/j.rmed.2016.10.017
Ito Y, Arita M, Kumagai S, Takei R, Noyama M, Tokioka F, Nishimura K, Koyama T, Notohara K, Ishida T (2017) Serological and morphological prognostic factors in patients with interstitial pneumonia with autoimmune features. BMC Pulm Med 17:111. https://doi.org/10.1186/s12890-017-0453-z
Collins B, Raghu G (2016) Interstitial pneumonia with autoimmune features: the new consensus-based definition for this cohort of patients should be broadened. Eur Respir J 47:1293–1295. https://doi.org/10.1183/13993003.02084-2015
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier J-F, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183:788–824. https://doi.org/10.1164/rccm.2009-040GL
Travis WD, Costabel U, Hansell DM, King T Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez Fernando J, Myers J, Protzko S, Raghu G, Richeldi L, Sverzellati N, Swigris J, Valeyre D (2013) An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 188:733–748. https://doi.org/10.1164/rccm.201308-1483ST
van den Hoogen F, Khanna D, Jaap F, Johnson Sindhu R, Murray B, Tyndall A, Matucci-Cerinic M, Naden RP, Medsger TA Jr, Carreira PE, Riemekasten G, Clements PJ, Denton CP, Distler O, Allanore Y, Furst DE, Gabrielli A, Mayes MD, van Laar JM, Seibold JR, Czirjak L, Steen VD, Inanc M, Kowal-Bielecka O, Muller-Ladner U, Valentini G, Veale DJ, Vonk MC, Walker UA, Chung L, Collier DH, Ellen CM, Fessler BJ, Guiducci S, Herrick A, Hsu VM, Jimenez S, Kahaleh B, Merkel PA, Sierakowski S, Silver RM, Simms RW, Varga J, Pope JE (2013) 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis 72:1747–1755. https://doi.org/10.1136/annrheumdis-2013-204424
Aletaha D, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JMW, Hobbs K, Huizinga TWJ, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Menard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovsky J, Wolfe F, Hawker G (2010) 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 69:1580–1588. https://doi.org/10.1136/ard.2010.138461
Bohan A, Peter JB (1975) Polymyositis and dermatomyositis (first of two parts). N Engl J Med 292:344–347. https://doi.org/10.1056/NEJM197502132920706
Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, Daniels TE, Fox PC, Fox RI, Kassan SS, Pillemer SR, Talal N, Weisman MH (2002) Classification criteria for Sjogren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 61:554–558. https://doi.org/10.1136/ard.61.6.554
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ (1982) The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25:1271–1277. https://doi.org/10.1002/art.1780251101
Kinder BW, Collard HR, Koth L, Daikh DI, Wolters PJ, Elicker B, Jones KD, King TE Jr (2007) Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med 176:691–697. https://doi.org/10.1164/rccm.200702-220OC
Danieli MG, Fraticelli P, Salvi A, Gabrielli A, Danieli G (1998) Undifferentiated connective tissue disease: natural history and evolution into definite CTD assessed in 84 patients initially diagnosed as early UCTD. Clin Rheumatol 17:195–201. https://doi.org/10.1007/BF01451046
Mosca M, Tani C, Neri C, Baldini C, Bombardieri S (2006) Undifferentiated connective tissue diseases (UCTD). Autoimmun Rev 6:1–4. https://doi.org/10.1016/j.autrev.2006.03.004
Strand MJ, Sprunger D, Cosgrove GP, Fernandez-Perez ER, Frankel SK, Huie TJ, Olson AL, Solomon J, Brown KK, Swigris JJ (2014) Pulmonary function and survival in idiopathic vs secondary usual interstitial pneumonia. Chest 146:775–785. https://doi.org/10.1378/chest.13-2388
Park JH, Kim DS, Park IN, Jang SJ, Kitaichi M, Nicholson Andrew G, Colby TV (2007) Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 175:705–711. https://doi.org/10.1164/rccm.200607-912OC
Fischer A, Swigris JJ, Bois Roland M, Lynch David A, Downey Gregory P, Cosgrove Gregory P, Frankel Stephen K, Fernandez-Perez Evans R, Gillis JoAnn Z, Brown Kevin K (2009) Anti-synthetase syndrome in ANA and anti-Jo-1 negative patients presenting with idiopathic interstitial pneumonia. Respir Med 103:1719–1724. https://doi.org/10.1016/j.rmed.2009.05.001
Funding
The study was supported by the National Natural Science Foundation of China (81570058), Jiangsu Provincial Medical Talent (ZDRCA2016058), and Jiangsu Social Development Project (BE2017604).
Author information
Authors and Affiliations
Contributions
Jinghong Dai and Hourong Cai contributed to the study design, data analysis and interpretation of HRCT, writing of the manuscript, and final approval; Lei Wang, Yan Xin, and Hui Li contributed to the clinical information collection, data analysis, and interpretation; Siyi Xu and Geyu Liang contributed to statistics analysis and interpretation; Kefeng Zhou and Jian He contributed to the interpretation of HRCT finding; and Fanqing Meng contributed to the histological assessment of the biopsy specimen.
Corresponding author
Ethics declarations
This was a retrospective study and the ethics approval was acquired in accordance with the policy of the Ethics Committee of the Affiliated Drum Tower Hospital of Nanjing University Medical School.
Disclosures
None.
Additional information
Summary at a glance
This is the first and large-scale population-based study to comprehensively investigate the clinical features and outcomes in Chinese patients with interstitial pneumonia with autoimmune features (IPAF). The current evidence suggested that IPAF had a distinct clinical characteristics compared with IPF and worse survival than non-IPF, but better than IPF.
Rights and permissions
About this article

Cite this article
Dai, J., Wang, L., Yan, X. et al. Clinical features, risk factors, and outcomes of patients with interstitial pneumonia with autoimmune features: a population-based study. Clin Rheumatol 37, 2125–2132 (2018). https://doi.org/10.1007/s10067-018-4111-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-018-4111-5