
Abstract
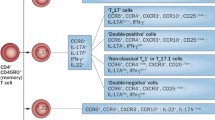
Th17 cells are a discrete subset of T cell subpopulation, which produce IL-17 and certain other pro-inflammatory cytokines. A regulatory role of Th17 cells have been proposed in several autoimmune diseases including psoriasis, psoriatic arthritis (PsA), ankylosing spondylitis (AS), rheumatoid arthritis, inflammatory bowel disease, systemic lupus erythematosus, and multiple sclerosis. Psoriatic disease is an autoimmune disease which mainly involves skin and joints. Until recently, psoriasis and PsA were thought to be Th1 mediated disease, but after the discovery of IL-17 and IL-17 knockout animal studies as well as human experimental data indicate a crucial role of the Th17 cells in the pathogenesis of psoriasis and PsA. Our research group have not only found abundance of CD4+IL-17+ T cells, mainly the memory phenotype (CD4RO+CD45RA−CD11a+) in the synovial fluid, but also have shown the existence of a functional IL-17 receptor in synovial fibroblast of psoriatic arthritis patients. Similarly, both animal and human studies indicate a regulatory role of the Th17 cells in AS; most critical observations are that Th17 cytokines (IL-17 and IL-22) can contribute to bone erosion, osteitis and new bone formation the hall mark skeletal features associated with the pathophysiology of AS. In this review article, we have discussed the contributing role of the IL-23/IL-17 axis in the pathogenesis of PsA and AS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spondyloarthritis (SpA) represents a group of interrelated inflammatory autoimmune diseases with common clinical features and a close association with HLA-B27. In SpA, ankylosing spondylitis (AS) and psoriatic arthritis (PsA) are most frequent. The hallmark of AS is acute and chronic spinal inflammation initiating in the sacroiliac joints, often coupled with enthesitis, presenting as chronic inflammation at the sites of ligamentous and tendinous insertions into bone [1]. Peripheral joint synovitis can be a prominent feature as well. PsA, often termed as “psoriatic disease” are autoimmune diseases which share certain similar pathological events and clinical features [2–5]. PsA is a heterogeneous disease characterized by involvement of skin, nails, peripheral and axial joints, and entheses; PsA develops in approximately 25 % of the psoriasis patients [6, 7].

Th17 in the pathogenesis of spondyloarthritis: Various external and internal cues trigger secretion of cytokines IL-23 TGF-β, IL-6, and IL-1 β during psoriatic disease and ankylosing spondylitis. These secreted cytokines can in turn participate in the generation of Th17 cells. These activated Th17 cells in the enthesis and in the synovial tissue can promote local inflammation and bone remodeling through a variety of effector mediators such as IL-17 and IL-22, which contribute to inflammation, bone erosion, and bone fusion
Although considerable progress has been made in deciphering the pathogenesis of both AS and PsA, the exact cause still remains a mystery. The role of innate and adaptive immune responses is now well established in their pathogenesis. Cytokines are the primary product of immune activation. Various cytokines play a detrimental role in initiation and progression of AS and PsA; however, understanding of their role in the disease is still evolving. In a complex milieu, pathogenic T cell subpopulations (Th1, Th9, Th17, Th22) and their signature cytokines (IFN-γ, TNF-α, IL-17, IL-22), chemokines, adhesion molecules, growth factors like nerve growth factor (NGF) and neuropeptides act in an integrated way through their corresponding receptors to determine the pathological events and tissue responses associated with various autoimmune rheumatologic diseases [1, 8–14].
A variety of genetic, immunological and environmental factors have been suggested to contribute to PsA pathogenesis [15, 16]. Single nucleotide polymorphisms in IL23A, IL23R as well as TRAF3IP2 (Act1), a downstream target of the IL-17 receptor (IL-17R), confer susceptibility to PsA, implying a central role of the IL-23/IL-17 axis in PsA disease pathogenesis [17–19]. Similarly, in addition to strong association with the MHC, a significant association has been confirmed between AS and single nucleotide polyphormisms (SNPs) in IL23R at chromosome 1p23 which indicates a major role for the interleukin (IL)-23 induced cytokine pathways in disease susceptibility of AS and has opened a novel outlook in the pathogenesis AS [20].
In a series of elegant experiments using mouse models and human tissues, it has been demonstrated that IL-23 induced Th17 cytokines (IL-17 and IL-22) can contribute to all four pathologic events in psoriatic arthritis: development of psoriatic plaque, pannus formation in the joint, joint erosion, and new bone formation. IL-23 activates the Th17 cells to produce Th17 cytokines such as IL-17 and IL-22. Th17 cell derived cytokines have more downstream effects such as epidermal thickening, synovial inflammation, angiogenesis, and cell trafficking [8, 12, 21, 22].
Considering the strong association of IL-23/IL-17 cytokine pathways, IL-17 targeted therapy for PsA and AS has been developed and preclinical and clinical trials so far have shown very encouraging results (Table 1) [23–26]. In this review article, we have discussed the contributing role of the IL-23/IL-17 axis in the disease processes of AS and PsA.
IL-17 and its receptor system
Following the discovery of the IL-17 encoding gene in a rodent T cell library in the early 90s, its human homologue with a cytokine-like activity was identified and labeled as IL-17 [27]. So far, IL-17A (commonly termed as IL-17) followed by IL-17F have been extensively studied amongst the six IL-17 family cytokines (IL-17A to IL-17F) [28]. IL-17A and IL-17F has 55 % amino acid resemblance and exists as homodimers sharing structural similarity having a disulfide bond and a cysteine-knot fold and sometimes as heterodimers IL-17A/F with inflammatory potential [29–31]. IL-17 receptor (IL-17R) complex is also multimeric analogous to IL-17. The earliest revealed subunit of IL-17R complex is IL-17RA, soon after other subunits of this complex have been recognized and termed as IL-17RB, IL-17RC, IL-17RD, and IL-17RE [32, 33]. Activation of intracellular signaling occurs through the binding of IL-17A and IL-17F to the receptor complex formed by IL-17RA and IL-17RC [33, 34]. Moreover, IL-17A also signals through a receptor complex of IL-17RA and IL-17RD [35]. Therefore, in the formation of functional IL-17R complex, IL-17RA is playing the pivotal role and might be a broad and thus a more potent therapeutic target compared to other ligands of IL-17 family.
IL-17 in psoriatic disease
Studies of human psoriatic lesions support a role for IL-12 and IL-23 in psoriasis. Several human studies have demonstrated increased levels of IL12p40 (shared by IL-12 and IL-23) messenger RNA (mRNA) in psoriatic lesions; further studies have demonstrated enrichment of psoriasis plaques with IL-23 specific p19 subunit [36–39]. Multiple studies have demonstrated increased expression of IL-17A, and IL-17F in psoriatic skin in contrast to the nonlesional psoriatic skin, and the up-regulation of IL-17A illustrated positive association with disease severity [40, 41]. Moreover, there is increased expression of the Th17 specific transcription factor (RORγt) and Th17 inducing cytokines (IL-23, IL-6, IL-1β) in lesional psoriatic skin versus nonlesional skin and skin of healthy volunteers [42]. Evidences also show overexpression of Th17 specific CC chemokine receptor, CCR6 and its ligand CCL20 in psoriatic skin lesions, and induction of CCL20 from keratinocytes by IL-17 [43]. All of these reports reflect the pivotal role of IL-17 in psoriasis. The major pathological roles of IL-17 in psoriasis are as follows: recruiting neutrophils to the epidermis of psoriatic lesion by increasing neutrophil specific chemokines [44, 45]; employing additional pathogenic Th17 cells by regulating CCL20 release from keratinocytes [43, 44]; stimulating expression of significant antimicrobial peptides of psoriasis like β-defensin, S100A7, S100A8, and S100A9, which consecutively act as pro-inflammatory stimulus [44, 46]; it also disrupts the skin barrier by downregulating expression of filaggrin and adhesion molecules in keratinocytes [47] and induces TNF-α release from dendritic cells and macrophages [26, 44].
Results of our study suggest that synovium of psoriatic arthritis is enriched with IL-17 producing CD4+ effector memory T cells and functionally active IL-17RA, the most well-recognized receptor for IL-17 [48]. Several reports suggest that IL-17 can influence bone and cartilage destruction in inflammatory arthritis [49, 50]. In animal arthritis model, disease severity is less in IL-17-deficient mice [51]. IL-17 receptor deficiency results in impaired synovial expression of IL-1 and MMP-3, MMP-9, and MMP-13 and prevents cartilage destruction during chronic reactivated streptococcal cell wall-induced arthritis [52]. To understand the role of IL-17 in the joint pathology of PsA, we examined the ability of IL-17 to induce MMP3 and cytokines by fibroblast-like synoviocytes (FLS) obtained from PsA synovium and have observed that FLS in PsA are tuned to a robust response with IL-17. There was a marked up-regulation of IL-6, IL-8, and MMP-3 upon exposure to IL-17 in cultured FLS from PsA patients [48]. IL-17 also promotes bone erosion through the up-regulation of receptor activator of nuclear factor kappa-B ligand (RANKL) [53, 54], a key regulator of osteoclastogenesis. Thus, the downstream effects of IL-17 are likely to influence all the major components of the pathologic events in skin and joint tissues of psoriatic disease.
IL-23 and IL-17 in the pathogenesis of AS
Ankylosing spondylitis (AS) is the prototypic feature of SpA. Ankylosing spondylitis is characterized by inflammation and progressive ankylosis of sacroiliac joints and the spine and is frequently associated with inflammation of the enthesis. TNF-α is considered to play a major role in driving inflammation in patients with AS, and TNF-α blocking agents provide marked improvement of pain/stiffness in AS patients. However, with time, it appears that TNF inhibition is not adequate to prevent the structural progression of AS. These clinical observations suggest regulatory role of other growth factors or inflammatory in the disease process of AS.
Knowledge of the involvement of the IL-23/Th17 pathway in SpA comes from limited clinical investigations. It has been reported that polymorphisms in the receptor for IL-23 are associated with ankylosing spondylitis and thus IL-23 and its receptor system likely to play a critical role in the disease susceptibility and the pathogenic of AS [55].
Serum concentrations of IL-23 and IL-17 have been found to be high in patients with AS [56]. Macrophages from patients with AS in response to lipopolysaccharide produce high levels of IL-23 [57]. IL-17 positive cells in AS facet joints also have been demonstrated [58]. In addition to AS and PsA, high levels of IL-17 in the synovial fluid of patients with reactive arthritis and undifferentiated SpA have been reported [59].
Simultaneous erosion and new bone formation the pathognomonic joint manifestations of spondyloartritis can be driven by cytokines of IL-23/IL-17 system
Bone resorption mediated by osteoclasts and new bone formation by osteoblasts is an integrated process in healthy bones. An elevated RANKL/osteoprotegerin ratio favors the differentiation of osteoclasts from monocytes and tips the balance towards bone resorption. Any disease model for AS and PsA remains incomplete if it does not provide explanations for bone resorption and new bone formation the hall mark features of bone changes in AS and PsA. Joint and bone tissues obtained from surgical samples of AS and PsA joints have demonstrated abundant osteoclasts at the pannus and bone junction [1, 60]. Also marked expression of RANKL and relatively faint staining of osteoprotegerin have been observed in the inflamed PsA synovium [60]. IL-17 promotes bone erosion through the up-regulation of RANKL [53, 54] a key regulator of osteoclastogenesis. Thus, IL-17 being a pro-inflammatory cytokine will act on synovial tissue of the joints of the spine/peripheral joints to induce the inflammatory/proliferatives cascades of pannus formation and in addition will promote joint erosion by its regulatory role on the osteoclasts.
Sherlock et al. in a passive transfer model of collagen-induced arthritis have reported that with IL-23 antibody treatment the development of pronounced axial and peripheral inflammation at the site of tendon and ligament attachment to bone could be ameliorated [61]. In this report, the investigators have also observed that injection of IL-23 minicircle in the B10.RIII mice could induce inflammation at regions adjacent to enthesis and also induced erosive arthritis along with new bone formation. A unique population of CD3+, IL-23R+, CD4−, CD8−, and RORγ T cells were identified in the enthesis. The unique CD3+, CD4−, and CD8− T cell phenotype did not arise in mice treated with an antibody to IL-23 [61]. The investigators have substantiated that IL-23 secreted by this unique CD3+, CD4−, and CD8− T cell subset was associated with pannus formation and joint erosion, presumably via up-regulation of TNF and IL-17; further, they demonstrated that in this model IL-22 induced new bone formation and apparently, IL-23 regulated the induction of IL-22.
So far, the existence and functions of the CD3+, CD4−, and CD8− resident T cell observed in mice remains unknown in humans. This encouraged us to investigate the source and the role of IL-22 in human spondyloarthritis. We observed that activated synovial T cells of PsA patients produce significantly more IL-22 compared to patients with OA; and PsA patients have higher concentration of IL-22 in their synovial fluid [23]. Further in this study we have observed that IL-22 is functionally active in PsA joints. Thus, it can be concluded that IL-23 induced Th17 cytokines (IL-17 and IL-22) can contribute to all four pathologic events in AS and PsA: development of psoriatic plaque, pannus formation in the joint, joint erosion, and new bone formation (Fig. 1) [21, 48, 61].
References
Appel H, Kuhne M, Spiekermann S, Ebhardt H, Grozdanovic Z, Köhler D, Dreimann M, Hempfing A, Rudwaleit M, Stein H, Metz-Stavenhagen P, Sieper J, Loddenkemper C (2006) Immunohistologic analysis of zygapophyseal joints in patients with ankylosing spondylitis. Arthritis Rheum 54(9):2845–51
Lowes MA, Bowcock AM, Krueger JG (2007) Pathogenesis and therapy of psoriasis. Nature 445(7130):866–73
Raychaudhuri SP (2013) A cutting edge overview: psoriatic disease. Clin Rev Allergy Immunol 44(2):109–13
Griffiths CE, Barker JN (2007) Pathogenesis and clinical features of psoriasis. Lancet 370(9583):263–71
Liu Y, Krueger JG, Bowcock AM (2007) Psoriasis: genetic associations and immune system changes. Genes Immun 8(1):1–12
Gladman DD, Farwell VT, Pellett F, Schentag C, Raham P (2003) HLA is a candidate region for psoriatic arthritis. evidence for excessive HLA sharing in sibling pairs. Hum Immunol 64(9):887–9
Amherd-Hoekstra A, Naher H, Lorenz HM, Enk AH (2010) Psoriatic arthritis: a review. J Dtsch Dermatol Ges 8(5):332–9
Raychaudhuri SP (2013) Role of IL-17 in psoriasis and psoriatic arthritis. Clin Rev Allergy Immunol 44:183–93
Raychaudhuri SP, Jiang WY, Raychaudhuri SK (2008) Revisiting the Koebner phenomenon: role of NGF and its receptor system in the pathogenesis of psoriasis. Am J Pathol 172(4):961–71
Raychaudhuri SP, Kundu-Raychaudhuri S, Tamura K, Masunaga T, Kubo K, Hanaoka K et al (2008) FR255734, a humanized, Fc-Silent, anti-CD28 antibody, improves psoriasis in the SCID mouse-psoriasis xenograft model. J Invest Dermatol 128(8):1969–76. doi:10.1038/jid.2008.38
Raychaudhuri SP, Mitra A, Datta Mitra A, Abria C, Raychaudhuri SK (2014) Th9 cells in inflammatory cascades of autoimmune arthritis. Arthritis Rheum 66(11):S708, Abstract 1602
Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG (2013) The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol 34(4):174–81
Kruithof E, Baeten D, Van den Bosch F, Mielants H, Veys EM, De Keyser F (2005) Histological evidence that infliximab treatment leads to downregulation of inflammation and tissue remodelling of the synovial membrane in spondyloarthropathy. Ann Rheum Dis 64:529–36
Raychaudhuri SP, Raychaudhuri SK (2004) Role of NGF and neurogenic inflammation in the pathogenesis of psoriasis. Prog Brain Res 146:433–7
Elder JT, Bruce AT, Gudjonsson JE, Johnston A, Stuart PE, Tejasvi T et al (2010) Molecular dissection of psoriasis: integrating genetics and biology. J Invest Dermatol 130(5):1213–26
Chandran V, Raychaudhuri SP (2010) Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis. J Autoimmun 34(3):J314–21
Bowes J, Orozco G, Flynn E, Ho P, Brier R, Marzo-Ortega H et al (2011) Confirmation of TNIP1 and IL23A as susceptibility loci for psoriatic arthritis. Ann Rheum Dis 70:1641–4
Filer C, Ho P, Smith RL, Griffiths C, Young HS, Worthington J et al (2012) Investigation of association of the IL12B and IL23R genes with psoriatic arthritis. Arthritis Rheum 64(4):1302
Huffmeier U, Uebe S, Ekici AB, Bowes J, Giardina E, Korendowych E et al (2010) Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat Genet 42:996–9
WTCCC & TASC (2007) Association scan of 14,500 nonsynonymous SNPs in four diseases identifiesautoimmunity variants. Nat Genet 39:1329–1337
Mitra A, Raychaudhuri SK, Raychaudhuri SP (2012) Functional role of IL-22 in psoriatic arthritis. Arthritis Res Ther 14(2):R65
Lories RJ, McInnes IB (2012) Primed for inflammation: enthesis-resident T cells. Nat Med 18(7):1018–9
Baeten D, Braun J, Baraliakos X et al (2014) Secukinumab, a monoclonal antibody to interleukin-17A, significantly improves signs and symptoms of active ankylosing spondylitis: results of a 52-week phase 3 randomized placebo-controlled trial with intravenous loading and subcutaneous maintenance dosing. Arthritis Rheum 66:S360, Abstract 819
Sieper J, Braun J, Baraliakos X et al (2014) Secukinumab, a monoclonal antibody to interleukin-17A, significantly improves signs and symptoms of active ankylosing spondylitis: results of a phase 3, randomized, placebo-controlled trial with subcutaneous loading and maintenance dosing. Arthritis Rheum 66:S232, Abstract 536
Mease PJ, Genovese MC, Greenwalk MW, Ritchlin CT, Beaulier AD, Deodhar A et al (2014) Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med 370(24):2295–306
Papp KA, Leonardi C, Menter A, Ortonne JP, Kreuger JG, Kricorian G et al (2012) Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 366(13):1181–9
Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK et al (1995) Human IL-17: a novel cytokine derived from T cells. J Immunol 155(12):5483–6
Tesmer LA, Lundy SK, Sarkar S, Fox DA (2008) Th17 cells in human disease. Immunol Rev 223:87–113
Kuestner RE, Taft DW, Haran A, Brandt CS, Brender T, Lum K et al (2007) Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17 F. J Immunol 179(8):5462–73
Liang SC, Long AJ, Bennett F, Whitters MJ, Karim R, Collins M et al (2007) An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol 179(11):7791–9
Hymowitz SG, Filvaroff EH, Jp Y, Lee J, Cai L, Risser P et al (2001) IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J 20(19):5332–41
Yao Z, Spriggs MK, Derry JM, Strockbine L, Park LS, Vanden Bos T et al (1997) Molecular characterization of the human interleukin (IL)-17 receptor. Cytokine 9(11):794–800
Ely LK, Fischer S, Garcia KC (2009) Structural basis of receptor sharing by interleukin 17 cytokines. Nat Immunol 10(12):1245–51
Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J et al (2006) Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol 177(1):36–9
Coimbra S, Figueriredo A, Castro E, Rocha-Pereira P, Santos-Silva A (2012) The roles of cells and cytokines in the pathogenesis of psoriasis. Int J Dermatol 51(4):389–95, quiz 395-8
Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F et al (2004) Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med 199(1):125–30
Piskin G, Sylva-Steenland R, Bos JD, Teunissen MB (2006) In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol 176(3):1908–15
Bovenschen HJ, van de Kerkhof PC, van Erp PE, Woestenenk R, Joosten I, Koenen HJ (2011) Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J Invest Dermatol 131(9):1853–60
Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein W, Mattson JD et al (2007) Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol 8(9):950–7
Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS et al (2008) Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol 128(5):1207–11
Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K (2009) Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol 160(2):319–24
van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD et al (2009) Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 182(9):5836–45
Homey B, Dieu-Nosjean MC, Wiesenborn A, Massacrier C, Pin JJ, Oldham E et al (2000) Up-regulation of macrophage inflammatory protein-3 alpha/CCL20 and CC chemokine receptor 6 in psoriasis. J Immunol 164(12):6621–32
Girolomoni G, Mrowietz U, Paul C (2012) Psoriasis: rationale for targeting interleukin-17. Br J Dermatol 167(4):717–24
Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan K, Suarex-Farinas M, Cardinale I et al (2008) Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol 159(5):1092–102
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M et al (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203(10):2271–9
Gutowska-Owsiak D, Schaupp AL, Salimi M, Selvakumar TA, McPherson T, Taylor S et al (2012) IL-17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp Dermatol 21(2):104–10
Raychaudhuri SP, Raychaudhuri SK, Genovese MC (2012) IL-17 receptor and its functional significance in psoriatic arthritis. Mol Cell Biochem 359(1-2):419–29
Chabaud M, Lubberts E, Joosten L, van den Berg W, Miossec P (2001) IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res 3:168–177
Koshy PJ, Henderson N, Logan C, Life PF, Cawston TE, Rowan AD (2002) IL-17 induces cartilage collagen breakdown: novel synergistic effects in combination with proinflammatory cytokines. Ann Rheum Dis 61(8):704–713
Nakae S, Nambu A, Sudo K, Iwakura Y (2003) Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol 171(11):6173–6177
Koenders MI, Kolls JK, Oppers-Walgreen B, van den Bersselaar L, Joosten LA, Schurr JR et al (2005) Interleukin-17 receptor deficiency results in impaired synovial expression of interleukin-1 and matrix metalloproteinases 3, 9, and 13 and prevents cartilage destruction during chronic reactivated Streptococcal cell wall induced arthritis. Arthritis Rheum 52(10):3239–3247
Koenders MI, Lubberts E, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Di Padova FE et al (2005) Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol 167(1):141–149
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S et al (1999) IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 103(9):1345–1352
Australo-Anglo-American Spondyloarthritis Consortium (TASC), Reveille JD, Sims AM, Danoy P, Evans DM, Leo P, Pointon JJ et al (2010) Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat Genet 42(2):123–127
Mei Y, Pan F, Gao J, Ge R, Duan Z, Zeng Z, Liao F, Xia G, Wang S, Xu S, Xu J, Zhang L, Ye D (2011) Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin Rheumatol 30:269–273
Zeng L, Lindstrom MJ, Smith JA (2011) Ankylosing spondylitis macrophage production of higher levels of interleukin-23 in response to lipopolysaccharide. Arthritis Rheum 63(12):3807–17
Appel H, Maier R, Wu P, Scheer R, Hempfing A, Kayser R, Thiel A, Radbruch A, Loddenkemper C, Sieper J (2011) Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res Ther 13:R95
Singh AK, Misra R, Aggarwal A (2011) Th17 associated cytokines in patients with reactive arthritis/undifferentiated spondylarthropathy. Clin Rheumatol 30:771–776
Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM (2003) Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest 111(6):821–31
Sherlock JP, Joyce-Shaikh B, Turner SP et al (2012) IL-23 induces spondyloarthropathy by acting on ROR-gammatþ CD3þCD4-CD8- entheseal resident T cells. Nat Med 18:1069–1076
Disclaimer
The views expressed in the article do not necessarily represent the views of the Department of Veterans Affairs or of the United States Government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Raychaudhuri, S.K., Saxena, A. & Raychaudhuri, S.P. Role of IL-17 in the pathogenesis of psoriatic arthritis and axial spondyloarthritis. Clin Rheumatol 34, 1019–1023 (2015). https://doi.org/10.1007/s10067-015-2961-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-015-2961-7