
Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare genetic inflammatory disorder characterized by progressive heterotopic ossification presenting as recurrent soft tissue masses and swelling which may cause disabling, restricted joint mobility. Congenital malformations of the hallux are characteristic features of classic FOP, predating the appearance of disabling features. As no definite treatment is available, the early diagnosis and prevention of exacerbating factors may lead to significant benefits in terms of the life quality of patients. A retrospective study of 12 consecutive FOP patients referred to and admitted in the rheumatology unit at an urban tertiary care academic center between 1991 and 2011. Data, such as age, gender, and past medical history, were collected from the medical history, physical examination, and skeletal survey in order to characterize the clinical presentations. All 12 children (six boys and six girls; ages 2.0–13.5 years) had congenital malformations of the great toes (microdactyly and hallux valgus deformity), in addition to heterotopic ossification presenting as multiple soft tissue tumor-like swellings. Spinal involvement, most notably in the cervical region, suggestive of an early FOP, was present in 83.3 %. Eleven patients (91.6 %) had a prior history of direct physical trauma, while 7 of 11 (63.6 %) had undergone invasive diagnostic procedures, both correlating with the exacerbations of their condition. Clinical awareness of fibrodysplasia ossificans progressiva and its early diagnostic features, particularly congenital malformations of the hallux, during a thorough neonatal examination may lead to an early diagnosis preventing the development of disabling, practically irreversible lesions of heterotopic ossification. Genetic and molecular studies can play a considerable role in the diagnosis of FOP in suspected cases. Early institution of prophylactic and precautionary measures, such as categorical avoidance of trauma and invasive procedures, can significantly reduce the debilitating acute exacerbations of the condition.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fibrodysplasia ossificans progressiva (FOP) is a rare genetic mesodermal inflammatory disorder of connective tissue. There are three known phenotypes: classical, FOP plus, and FOP variant. A common recurrent mutation in the ACVR1 gene has been described and is usually sporadically acquired in children of previously unaffected families. Inherited forms are in an autosomal dominant fashion, with complete penetration and variable gene expressivity. Although the exact pathogenesis is unclear, recent genome-wide association studies (GWAS) have mapped chromosomal bands 4q27-314 and 17q21-22 as target gene locations. Trauma has been reported as an initiating and exacerbating factor, however, rarely been appreciated [8].
The incidence of FOP worldwide has been reported to be approximately 1 per 2 million of the population with the most cases found in the USA, which may indicate the importance of routine examination during the neonatal and early infancy period in attaining the diagnosis [8].
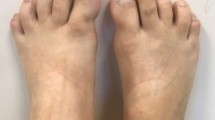
The earliest recognizable clinical features of classical FOP detectable at birth are great toes malformations, hallux valgus deformity, and microdactyly. The thumbs are also occasionally involved. Other manifestations, such as soft tissue tumor lesions may develop subsequently involving the axial region initially. The painful swelling of connective tissues is followed by progressive fibrosis, calcification, and eventually, true ossification.
Clinical features sufficient to make the diagnosis of classical FOP include congenital malformations of the hallux, which are usually symmetrical congenital malformations of the distal extremities [1–4, 7, 9], with recurrent externally triggered flare-ups leading to cumulative disability and joint immobilization [8]. Clinical knowledge and awareness is of paramount importance in the early diagnosis of FOP as imaging findings, such as ossification and calcification, are inconclusive and will appear at later and irreversible stages. Histological examination is also inconclusive with frequent misdiagnosis as osteosarcoma [1].
In underdeveloped countries where the cost of diagnostic procedures is expensive and not easily accessible, the clinical suspicion based on great toe findings alone can be sufficient in assisting the initial diagnosis. Subsequent genetic testing to confirm the diagnosis can later be arranged prior to heterotopic ossification (HO) and appearance of severe restricted joint mobility. Due to unawareness and poor clinical suspicion, the rate of misdiagnosis and delay in diagnosis is high, nearly 90 % worldwide was misdiagnosed, and 67 % undergone unnecessary diagnostic procedures leading to permanent injury and lifelong disability in more than 50 % [8]. The most common incorrect diagnoses include malignancy, aggressive juvenile fibromatosis, and desmoids tumor [8].
FOP has a relapsing and remitting nature resulting in restricted joint mobility and reduced life expectancy. Exacerbating factors can be self-inflicted, accidental, as a result of trauma, muscle fatigue, viral infection, or iatrogenic such as diagnostic biopsies and invasive surgical procedures. Most patients are wheelchair-bound by the age of 30, with an average life expectancy of 50–60 years as a result of frequent thoracic complications. Also, there is a huge burden on their families, both emotionally and financially.
Method
A retrospective study was conducted at Imam Khomeini Hospital, a metropolitan tertiary care academic center. Data from all 12 consecutive children diagnosed with FOP after being referred to the Pediatric Rheumatology Department between November 1983 and December 2008, were analyzed. A single pediatrician was involved in the diagnosis and follow-up of all the cases. Detailed medical history obtained and physical examination findings were clearly documented, and skeletal surveys were ordered.
The diagnostic criteria included congenital skeletal malformations of the great toes and calcification and progressive heterotopic ossification in characteristic anatomic distributions such as the neck, back, trunk, and extremities. Data included gender, age, and age of onset; the delay in diagnosis; the initial anatomic region of the onset and current regions of impaired joint mobility; past medical history of trauma; previous family history of the condition; original findings and congenital malformations on clinical examination; radiographic findings; and response to treatment
Studies were approved by the Tehran University Hospital’s Ethics Committee Review Board on human research and informed written parental consent obtained. The questionnaires were filled in by the medical team, and the collected data were analyzed with SPSS software (ver. 14).
Results
A total of 12 children, 6 boys (50 %) and 6 girls (50 %), were diagnosed to have FOP with an age range of 2.0–13.5 years. The mean age at the time of diagnosis was 7 years old, and the mean delay from the onset of symptoms to diagnosis was 22 months (range, 1 month–8.1 years). In all cases where a biopsy was performed, confounding differentials on histopathology specimens included systemic sclerosis, chronic juvenile myositis, or malignancy.
In 11 cases who presented with reduced joint mobility, the most common initial sites of involvement, in descending order of frequency, were: the cervical region (50 %), shoulder girdle, upper extremities, thoracic and lumbar spine, hip, and lower extremities. The only patient with no restricted range of motion was a 3-year-old girl diagnosed within 6 months of an initial presentation of a suspicious mass on the forehead. Eleven (91.7 %) patients had a history of direct physical trauma, and seven patients (58.3 %) had a history of invasive biopsy at sites of HO, which both resulted in exacerbation of the condition. None of the children had a family history of FOP (Tables 1 and 2).
Malformations of the great toe were present in all the cases at birth. Solid tumor-like masses were also present in all patients and appeared during the course of the disease. Torticollis was observed in eight patients (66.7 %); deformities of the thorax deformities, in seven (58.3 %); and scoliosis, in four children (33.3 %). Other findings included malformations of the fingers such as clinodactyly or microdactyly in eight patients (66.7 %); mental retardation, in one case (8.3 %); absence of incisors, in two cases (16.7 %); and a failure of physical growth and developmental skeletal anomalies, in three cases (25 %). None of the cases suffered from hearing problems or alopecia (Table 3).
In addition to symmetrical radiographic changes in the great toes present in all the patients, 11 (91.7 %) had signs of soft tissue calcification, 4 (33.3 %) had hip involvement, 6 (50 %) had evidence of cervical ankylosis, 3 (25 %) had multiple exostoses in the tibia, and 3 cases (25 %) had short and broad femoral neck (Table 4).
Nine patients received regular follow-up and therapeutic intervention in the form of alendronate (5–10 mg; once daily), corticosteroids, and nonsteroidal anti-inflammatory drugs (NSAIDs). The average duration of follow-up was between 1 and 10 years, with significant reduction in swelling, joint movement restriction, and the episodes of acute exacerbations and less new soft tissue lesions noted.
Discussion
FOP is a genetic disorder of progressive soft tissue ossification, which is usually the result of spontaneous de novo mutations, although familial cases have been described. Initial presentation, which is usually in the first two decades of life, involves recurrent episodes of painful soft tissue swelling including muscles; fascia and aponeuroses are involved, but subcutaneous tissues are spared—a distinguishing feature from progressive osseous heteroplasia (POH). The later calcification and true endochondral ossification result in characteristic lesions of heterotopic ossification. Congenital malformations of the digits, particularly the great toes, are important diagnostic features present from birth in all cases of classic FOP to date [1, 3]. Factors, such as gender [4] and race, are believed to have no bearing on the epidemiology of the FOP, though increasing paternal age could have a causative effect.
Although definite diagnosis of classical FOP can be made predominantly on clinical grounds, radiographic findings may assist in the diagnosis in cases where heterotopic ossification is less obvious. With the advent of the discovery of the gene responsible and the recurrent FOP mutation in ACRV1, molecular genetic testing can provide a definitive evidence in children with preosseous or even no signs of heterotopic ossification where the differential is most perplexing [1, 2, 4]. These mapping techniques are among the most specific in clinical genetics [1, 5]. The exact etiology is unknown, but the main predisposing protein is believed to be the bone morphogenic protein (BMP4), which signals endochondral ossification via the AKP pathway.
None of the patients had a family history of FOP, suggesting that they had acquired the more prevalent sporadic form described in most other studies [1, 2, 5]. Trauma is a trigger factor in most cases, frequently in conjunction with ensuing iatrogenic and confounding biopsies of solid tissue lesions. The invasive investigations not only contributed to exacerbations of FOP and the progressive disability, but were also inconclusive due to the nonspecific histological evidence observed. Other reported associations with exacerbation include muscle injections, viral illness, and even muscle fatigue [2–4, 8]. It is believed that although endochondral ossification is not subject to aberrant regulation in FOP, initiation does not occur without an initial inflammatory trigger.
All patients had congenital malformations of the great toes in the form of microdactyly and a hallux valgus deformity which is characteristic and sufficient to diagnose “classic” FOP when presented in conjunction with the evidence of heterotopic ossification [5]. Presence of FOP signs at birth is due to possible phenotypic effects in utero and is probably the most useful feature in facilitating early diagnosis [2, 3, 5]. Indeed, suspicion based on malformations of the hallux and preosseous soft tissue lesions can justify genetic testing to confirm the diagnosis prior to and without the previously necessary confirmation of HO [5]. In our study, the presence of solid tumor-like lumps was a sufficient evidence of HO to confirm the diagnosis without the need to expensive genetic testing. Consistent with previous studies, solid-like tumors were more pronounced in sites with proceeding direct trauma or invasive biopsies [2–4, 9].
Restricted movement and joint immobilization are signs of late progressive disease. The severity of debilitation heavily depends on the time of diagnosis. Similar to previous reports, in our study where the diagnosis was made at a relatively early stage, patients experienced problems predominantly in the axial skeleton, in regions such as the neck, back, and lumbar spine [2, 3]. Shore et al. [9] concluded that the progression of FOP tended to begin in the shoulder girdle, spreading to the vertebra, and finally the hip. Also, in a study by Shore et al. [10], the axial shoulder involvement appeared to occur earlier than distal extremities.
Other features associated with more progressive and advanced disease included involvement of the limbs and vertebrae, torticollis, deformities of the thorax, scoliosis, and malformations of the fingers such as clinodactyly of the fifth finger and microdactyly of the thumb. Skeletal malformations of the hand [11] and kyphoscoliosis [3] have both been previously reported. Other findings in our study, previously described by others, included one case with mental retardation, two children with absent upper incisors, and three children with a failure of physical growth. Other findings, such as hearing loss and alopecia, were not evident in our study.
One previously unreported finding presented in four of our patients was an increased length of the second toe, both relative to the other toes on the same extremity and the normal size in the population. This may, however, merely be a common anatomic variation or simply an illusion due to the relative short length of the adjacent hallux.
Radiographic findings prior to invasive interventions can assist in confirming clinical diagnosis without causing iatrogenic harm in cases where swelling has progressed to calcification. The universal changes in the hallux observed in these 12 patients have also been reported in all the other cases of classic FOP [3, 4], as well as the correlation between the presence, number, and severity of tumor-like lesions of HO with the time of diagnosis [4].
Bony ankylosis of the cervical vertebra was present in six patients; exostoses were present in three patients; shortening and broadening of femoral neck was seen in three patients. These findings, together with others such as ossification of paraspinal muscles [12] and potential formation of bridges between adjacent islands of heterotopic ossification have been reported in many other cases as contributors to the severe restriction in joint mobility observed [4, 7].
Nine patients, still subject to regular follow-up at the time of writing, were receiving bisphonates (alendronate 5–10 mg; once daily) and low-dose corticosteroids and NSAIDs, in cases where they had active inflammation. Three cases were unfortunately lost and not followed up.
Those children who together with their families were adequately educated about their predicament, the necessity of avoiding any form of trauma, and full compliance with the medication, in which they seemed to experience fewer flare-ups; even when presented, these tended to be milder and more responsive to the treatment. Prophylaxis is of significant importance given the absence of curative treatment to date [7, 10, 14], although the benefit of regimens with bisphosphonates and steroids in reducing flare-up has been highlighted [2, 13]. Some studies showed that continuous cycles of sodium etidronate (5–10 mg; PO) for 2 to 3 months, together with the long-term cessation of steroid and NSAID use, improve physical signs and can result in the cessation of heterotopic ossification[6]. Management guidelines currently focus on early diagnosis, prevention of trauma, and avoidance of invasive diagnostic tests [5, 7, 9]. Molecular treatment based on the mechanism of the gene involved will be the future goal in treating the sufferer from FOP [2].
The mean delay time in the diagnosis is mostly due to the rarity of FOP and the absence of sufficient clinical recognition of the condition by clinicians including the insufficient coverage in recognized medical texts [8]. Our data is consistent with previous reports that nearly 90 % of FOP patients worldwide are initially misdiagnosed, and that 67 % undergo avoidable procedures that have led to permanent harm and life-long disability in more than 50 % of patients.
Conclusions
Significance of:
-
Careful attention to clinical findings such as congenital malformations of the hallux and solid osseous and tumor-like lumps,
-
Avoidance of biopsy and other invasive diagnostic tests, and
-
Preventing any type of trauma
Appreciation of these points can considerably improve the quality of life of patients by delaying, reducing, or even preventing joint mobility restriction; otherwise, it could lead to physically and emotionally crippling irreversible lifelong disability.
References
Cohen RB, Hahn GV, Tabas J et al (1993) The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. J Bone Joint Surg 75(2):215–219
Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS (1994) Age and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Rel Res 301:243–248
Kaplan FS, Strear CM, Zasloff MA (1994) Radiographic and scintigraphic features of modeling and remodeling in the heterotopic skeleton of patients who have fibrodysplasia ossificans progressiva. Clin Orthop Rel Res 304:238–247
Mahboubi S, Glaser DL, Shore EM, Kaplan FS (2001) Fibrodysplasia ossificans progressiva. Pediatr Radiol 31(5):307–314
Kaplan FS, Glaser DL, Shore EM et al (2005) The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab 3(3–4):183–188
Kaplan FS, Glaser DL (2005) Thoracic insufficiency syndrome in patients with fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab 3(3–4):213–216
Kaplan FS, Tabas J, Gannon FH, Finkel G, Hahn GV, Zasloff MA (1993) The histopathology of fibrodysplasia ossificans progressiva: an endochondral process. J Bone Joint Surg 75(2):220–230
Kitterman JA, Kantanie S, Rocke DM, Kaplan FS (2005) Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 116(5):e654–e661. http://www.pediatrics.org/cgi/content/full/116/5/e654
Shore EM, Feldman GJ, Xu M, Kaplan FS (2005) The genetics of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab 3(3–4):201–204
Shore EM, Xu M, Feldman GJ et al (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38(5):525–527
Schaffer AA, Kaplan FS, Tracy MR et al (2005) Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel–Feil syndrome. Spine 30(12):1379–1385
Pignolo RJ, Suda RK, Kaplan FS (2005) The fibrodysplasia ossificans progressiva lesion. Clin Rev Bone Miner Metab 3(3–4):195–200
Ravitsky V (2006) Disclosing individual genetic results to research participants. Am J Bioethics 6(6):8–17
Pelias MK (2006) Genetic testing of children for adult-onset diseases: is testing in the child’s best interest? Mt Sinai J Med 73(3):605–608
Acknowledgments
In writing this paper, data was used from Dr. A. Tofighi-Zavareh, our dear colleague. We are also thankful to the medical records staff at Imam Khomeini hospital.
Disclosures
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Raees-Karami, S.R., Jafarieh, H., Ziyayi, V. et al. Evaluation of 20 years experience of fibrodysplasia ossificans progressiva in Iran: lessons for early diagnosis and prevention. Clin Rheumatol 31, 1133–1137 (2012). https://doi.org/10.1007/s10067-012-1968-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-012-1968-6