
Abstract
This study aims to report the concomitant diseases observed and damage outcome in a cohort of patients with adult idiopathic inflammatory myositis (IIM) during long-term follow-up. All patients with IIM were identified from a single centre (follow-up between 1979 and 2006) and fulfilled at least three of the four Bohan and Peter criteria. Patients with inclusion body myositis, juvenile-onset myositis and overt overlap syndromes were excluded. Medical notes were retrospectively reviewed. Concomitant diseases identified were divided into 12 different organ systems (bone, cardiac, respiratory, gastrointestinal, renal, central nervous, malignancy, infection, endocrine, eyes, dermatological and haematological). Patient damage index was calculated using the Myositis Damage Index tool. Fifty-five patients (31 polymyositis, 24 dermatomyositis) were identified. The most prevalent organ system involved was lung with 40 events per 1,000 patient years follow-up. There was significant steroid-related complications with 17/18 patients with bone involvement having osteopenia/osteoporosis. Sjogren's syndrome (n = 3) was the most frequent concomitant auto-immune disease observed. Patients with a higher number of organ systems involved had a significantly higher damage index (r = 0.48, p = 0.001). White patients showed a significant trend to develop more than three other organ system involvement (p < 0.0001) and myositis-related lung disease (p < 0.0001) compared to other races. There is significant steroid-related morbidity in adult IIM patients under long-term follow-up. The prevalence of another concomitant auto-immune disease unlike patients with lupus or Sjogren's syndrome is low.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Polymyositis (PM) and dermatomyositis (DM) are idiopathic inflammatory myopathies (IIM) characterised clinically by symmetrical muscle weakness and pathologically by muscle inflammation. Systemic manifestations of other organ/systems are often involved contributing to substantial morbidity and mortality.
Previous studies have demonstrated significant morbidity in these patients related to the underlying IIM using muscle strength testing and functional outcome assessments [1–4]. The 5-year survival rates for these patients range from 63% to 95% with the primary causes of death related to cardiac, respiratory and cancer involvement depending on the population studied [5–7].
A number of reports have described associated clinical features and outcome of patients with IIM [8–10]. Patients with IIM are typically treated with immunosuppressives and long-term steroid therapy. The development of other concomitant diseases, which may or may not be related, to the underlying myositis is not known. We have previously reviewed the rates of other auto-immune rheumatic diseases concordance in our cohort of patients with systemic lupus erythematosus (SLE) [11, 12] and Sjogren's syndrome [13]. In both cases, rates of approximately 30% were noted and we wondered if the same level of disease concordance was evident in our patients with myositis.
This study was undertaken to determine the concomitant diseases (including other auto-immune rheumatic diseases) that occurred in a cohort of IIM patients that has been followed up over a 27-year period. We also determined whether the level of damage caused by both myositis and its treatment was associated with the development of other organ systems involved.
Materials and methods
Patients
All patients with the diagnosis of IIM were identified from the University College London (UCL) myositis database. Patients with inclusion body myositis and juvenile myositis were excluded from analysis including patients whose myositis clearly occurred in the context of another auto-immune rheumatic disease notably systemic lupus erythematosus. This cohort has been followed up from 1979 to 2006. All patients fulfilled at least three of the four Bohan and Peter criteria [14, 15] for the diagnosis of myositis. Medical charts were retrospectively reviewed for demographics, disease duration, clinical features and immunosuppressive use. The pattern of myositis disease was noted and sub-classified to monophasic disease (single episode of active disease), relapsing–remitting disease (disease flares associated with disease-free intervals) and chronic progressive disease (ongoing active disease despite treatment).
Concomitant diseases (related and unrelated to the underlying IIM) after the diagnosis of IIM were analysed in detail. The concomitant medical diseases identified were divided into 12 different organ systems: bone, cardiac, respiratory, gastrointestinal, renal, central nervous, malignancy, infection, eyes, dermatological, endocrine and haematological. Concomitant auto-immune diseases that developed after the diagnosis of IIM were also identified. The diagnosis of Sjogren's syndrome was made according to the European classification criteria of 1993 [16]. The diagnosis of an additional condition was made clinically by the attending rheumatologist with the relevant confirmatory investigations or referral to another speciality where appropriate when clinical suspicion was raised. Patients who were established on more than 6 months of steroid therapy underwent a bone densitometry scan to confirm a diagnosis of osteopenia/osteoporosis. The average daily corticosteroid doses were calculated for each patient based on the cumulative and total duration of corticosteroid use.
Laboratory analysis
All patients had bloods for serum creatine kinase (CK; units per litre) and autoantibody screening performed, i.e. antinuclear antibodies (ANA) by immunofluorescence and antibodies to the extractable nuclear antigens (ENA) including the myositis specific antibody (MSA), anti-Jo1 antibody, by enzyme-linked immunosorbent assay (Shield Diagnostics, Dundee, UK).
Assessment of myositis damage
A damage index for each patient was calculated using the validated tool, Myositis Damage Index (MDI) on chart review. This tool, modified from the Systemic Lupus International Collaborative Clinics/American College of Rheumatology Damage index, is used to assess the extent and severity of damage developing in 11 different organ systems. The MDI extent of damage score (range 0–38) was calculated for all patients in this study.
Statistical analysis
The Prism and SPSS statistical software programmes were utilised for data analysis. The prevalence of each organ system was measured as number of events per 1,000 patient years follow-up. Chi-squared or Fisher's exact analysis was used to compare characteristics of patients with a particular organ system involvement and those who did not. Kaplan–Meier curves were generated to analyse time to development of osteopenia/osteoporosis according to steroid doses, time to development of more than three-organ-system involvement and lung disease according to various ethnic groups. The association between the number of organ system involvement and damage index was analysed using regression analysis with Spearman's correlation coefficient. A p value less than 0.05 was regarded as statistically significant.
Results
Fifty-five patients with IIM were identified from the UCL database. Thirty-one patients had PM and the remaining 24 had DM. As expected, there was a female preponderance with a female-to-male ratio of 2:1. The mean age of diagnosis was 41 years (range 16–65) and the median follow-up duration was 9 years (range 4 months–35 years). Eighty-seven percent (n = 48) of the patients had a muscle biopsy to establish the diagnosis of IIM. The majority ethnic group was white (n = 33, 60%) and the remaining groups were blacks (n = 8, 15%), Asians (n = 4, 7%) and other (n = 10, 18%). Most of the patients had chronic progressive myositis disease pattern (22/55, 40%) followed by monophasic (n = 17, 33%) and relapsing and remitting disease (n = 16, 29%).
The mortality rate in our series was 11%. Six patients in the cohort have died and half of these patients died from pulmonary complications. Causes of death include pneumonia (n = 2), pulmonary hypertension, trauma-related head injury, hypertrophic obstructive cardiomyopathy and ruptured abdominal viscus. Four of the patients who died had chronic progressive myositis disease.
The majority of patients presented with upper (n = 53, 96%) and lower limb (n = 49, 89%) weakness. Half of the patients (n = 28, 51%) had associated myalgia. Joint involvement was common with 24 patients (44%) presenting with arthralgia/arthritis. Constitutional features of fever, weight loss and fatigue were also common with 21 patients (38%) having these symptoms. Respiratory and bulbar muscle weakness was uncommon features with 4% (n = 2) and 7% (n = 4) of our cohort presenting with these symptoms, respectively.
The mean peak CK was 4,167 IU/L with 85% (n = 47) of patients presenting with an elevated CK.
Of the 55 patients, 27 (49%) tested positive for ANA. Ten patients (18%) had the presence of anti-Jo1 antibody. The majority of the patients with anti-Jo1 antibodies (70%) had underlying lung disease related to myositis. Other auto-antibodies [rheumatoid factor (n = 4), RNP (n = 3), Ro (n = 5) and La (n = 2)] were present only in a minority of patients.
Azathioprine and methotrexate were the most common disease-modifying anti-rheumatic drugs used in our cohort for the treatment of IIM and all patients were established on corticosteroid therapy. Eighteen patients have had intravenous immunoglobulins and four patients were trialed on rituximab (anti-CD20 monoclonal antibody) due to refractory disease.
The prevalence and types of concomitant diseases after the diagnosis of IIM are shown in Table 1. Lung disease was the most prevalent organ system with 40 events per 1,000 patient years follow-up (n = 20). All but one patient had lung involvement related to myositis with interstitial lung disease (ILD). The predominant pattern of ILD was nonspecific interstitial pneumonia (n = 13) whilst the remaining three patients had organising pneumonia.
Pulmonary hypertension was observed in 7% (4/55) of patients. Interestingly, one DM patient had isolated pulmonary hypertension with no evidence of associated pulmonary fibrosis.
There were a significant number of patients with metabolic bone disease (n = 18), in particular, osteoporosis and osteopenia related to steroid treatment for IIM (17/18). In addition, other steroid-related complications were observed with the majority of patients with eye involvement diagnosed with cataracts (6/8–75%) and three of the five diabetic patients identified in the endocrine system had steroid-induced diabetes.
Only one patient had myositis-related cardiac involvement with an underlying cardiomyopathy. The main gastrointestinal diseases observed were gastritis (four of eight) and fatty liver (three of eight).
Five patients had malignancies and one patient had two malignancies diagnosed (lung and lymphoma). All cancers occurred at a median time of 14 years after the diagnosis of myositis except for one patient where endometrial carcinoma was diagnosed shortly after she presented with DM.
A third of the serious infections observed (6/16—38%) were opportunistic in nature. Four patients (25%) had pneumonia. Half of the patients with neurological organ system involvement (five of ten) had carpal tunnel syndrome. Nine patients (16%) had renal disease with microscopic haematuria. Two patients had previous acute renal failure. One patient recovered from acute renal failure and the other presented with haemolytic uraemic syndrome (HUS) complicated with renal failure at initial presentation of her myositis.
Skin disease not related to IIM was present in 12 patients (22%) and nearly half (5/12) of these patients had seborrhoeic dermatitis. The majority of patients with haematological involvement (6/10—60%) had an underlying monoclonal gammopathy.
Sjogren’s syndrome (n = 3, 6%) was the most frequent concomitant auto-immune disease observed. Other auto-immune diseases observed were immune-mediated hypothyroidism (n = 2), auto-immune thrombocytopenia (n = 1) and coeliac disease (n = 1).
We then determined if there were any associated clinical or serological features (ANA and ENA antibodies) that might distinguish patients with a particular organ involvement versus patients who did not have the particular organ involved. Twelve clinical features were analysed: muscle weakness, DM rash, respiratory muscle weakness, bulbar muscle weakness, myalgia, arthritis, distal ulceration, calcinosis, Raynaud’s phenomena, fatigue, fever and weight loss. Significant results were found for five organ systems—lung, cardiac, endocrine, infection and skin (Table 2).
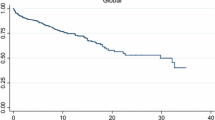
Thirteen patients had to be excluded from the corticosteroid analysis as the data collection was incomplete. Patients were divided to two groups—with or without osteoporosis/osteopenia from the remaining 42 patients. The average daily steroid dose for each group was 8.3 and 13.5 mg, respectively. Patients with long-term steroid doses of more than 5 mg had a significantly shorter time to develop osteoporosis/osteopenia (p < 0.0001, Fig. 1).

Time to development of osteoporosis/osteopenia (high- versus low-dose prednisolone)
We also found ethnic differences in our cohort where white patients showed significant trend to developing myositis-related lung disease and ≥3-organ-system involvement (p < 0.001) compared to other races (Figs. 2 and 3).

Time to development of ≥3-organ-system involvement and ethnicity

Time to development of lung disease and ethnicity
Discussion
This study has shown that the presence of concomitant diseases and therapy contributes to significant co-morbidities in patients with IIM. We have shown, on long-term follow-up, that there is significant morbidity related to corticosteroid use, principally osteoporosis and osteopenia. Ninety-four percent of patients with bone organ involvement developed osteoporosis/osteopenia in this study. Furthermore, other complications related to long-term corticosteroid use, i.e. cataracts and steroid-induced diabetes, were also observed. It is worth noting that our patients have been followed up since 1979 and specific treatments to prevent osteoporosis, specifically, bisphosphonates, were not available until 9 years ago. This may explain the high prevalence of osteoporosis/osteopenia in this series. In our cohort, patients treated with an average daily steroid dose of more than 5 mg had a significantly shorter time to develop osteoporosis. This finding highlights the importance of tapering steroid doses to the lowest possible dose with early introduction of a steroid-sparing agent whilst maintaining the IIM in remission. Corticosteroid-related morbidity has been reported to significantly contribute to functional disability measured by the Health Assessment Questionnaire [17].
The mortality rate in our cohort was 11% which is comparable to other studies that have estimated disease-related deaths in 10–22% of patients [3, 18].
Almost all the patients with lung involvement had lung disease related to myositis (18/19, 33% of the whole cohort). Our results are similar to previous studies of reported IIM-associated interstitial lung disease occurring in 23.1–39% of patients [19, 20]. Of the 55 patients, four patients developed pulmonary hypertension in this study. In the study by Marie et al., of the three patients who died in a cohort of 36 PM/DM patients with ILD, one developed pulmonary hypertension 3 years after the occurrence of ILD. Interestingly, there was one patient (DM) in our cohort with confirmed isolated pulmonary hypertension on echocardiograph and cardiac catheterisation without underlying ILD. Although two cases of pulmonary hypertension in dermatomyositis have been reported [21, 22], the incidence of isolated pulmonary hypertension in IIM is not known. The possible association of pulmonary HT in dermatomyositis may be explained by its pathogenesis where DM is generally regarded as a vasculopathy compared to PM where the inflammatory infiltrates are predominantly within the muscle fibres [23].
The risk of cancer in patients with DM and PM is well documented [24–26]. This risk has been reported to be considerably higher in DM compared to PM. The majority of cancers are diagnosed within 2 years of diagnosis of the underlying IIM. Five patients in this study had malignancies and all cancers (except one) occurred many years after the diagnosis of IIM with a median time of 14 years. These results emphasise the importance of ongoing vigilance for the clinician in managing IIM patients as malignancies can occur many years after the initial myositis diagnosis. A recent study investigating the incidence of malignancies in the SLE population showed that the majority of the cancers occurred more than a year after the diagnosis of SLE [27]. Other factors such as exposure to long-term immunosuppressive use have been reported to be associated with the risk of haematological malignancy in the rheumatoid arthritis population [28].
Previous studies have shown that about 30% of patients with SLE and Sjogren's syndrome developed another auto-immune rheumatic disease [11–13]. We found a much lower prevalence (6%) of another auto-immune rheumatic disease in our myositis series when overt and well-established overlap syndromes were excluded. Sjogren's syndrome was the most frequent concomitant auto-immune disease identified. Two patients developed auto-immune-mediated hypothyroidism. Auto-immune thyroid diseases such as Hashimoto's and Graves diseases are found to be more common amongst patients with an underlying systemic auto-immune disease [29]. A study by Koh et al. reported that SLE was the commonest associated auto-immune rheumatic disease in their cohort of PM/DM patients that were sub-classified with another connective tissue disorder [30]. Clustering of auto-immune diseases has been recognised in familial studies suggesting a genetic basis for the development of these conditions [31].
Of the patients who developed serious infections, a third was opportunistic in nature. Opportunistic infections have been reported in 11.5–21% of patients with PM/DM [32, 33] and one of these studies demonstrated an associated high mortality rate of 27.7% [32]. One patient in our cohort with underlying myositis ILD died from pneumonia. This highlights the need for early recognition and prompt treatment of infections in this population, in particular, atypical and opportunistic infections.
One patient had HUS at initial presentation of her muscle disease. This is an uncommon association and one reported case of HUS in a patient with PM has been described [34].
The International Myositis and Clinical Studies Group group has attempted to develop a consensus to develop and validate outcome measures to describe myositis disease activity and damage for use in clinical trials [35]. The myositis intention to treat index and myositis disease activity assessment visual analogue scale which aim to capture activity in patients with IIM have recently been validated [36, 37]. The MDI assesses the extent and severity of damage in different organ systems. This tool assesses the presence of damage for more than 6 months which is potentially irreversible. We found a significant correlation with the number of organ systems involved and the damage index measured in our patient cohort. Patients who had more than three other organ systems involved had a higher damage index. This is, perhaps, not a surprising finding and emphasises the importance of treating active IIM before irreversible damage occurs.
Interestingly, we found that white patients showed a significant trend to develop myositis-related lung disease and a higher number (≥3) of other organ systems involved compared to other races implying that ethnic variation may play a role in the clinical presentation of IIM. This variation may be related to immunogenetic factors. Shamim et al. have reported a difference of prevalence rates in clinical and serologic features depending on ethnic groups [38]. Furthermore, a recent study demonstrated that African American IIM patients shared distinct immunogenetic susceptibility factors compared to European American patients [39].
There are a number of limitations of this study which include the retrospective design, lack of a control group and absence of a standardised specific follow-up protocol to detect a particular concomitant disease.
Nonetheless, this study highlights the high morbidity associated with IIM, in particular, steroid-related complications on long-term follow-up. We recommend the lowest steroid dose whenever possible in controlling IIM. Furthermore, this study reinforces ongoing vigilance for an underlying malignancy which can present later in the course of disease. In addition, we found a much lower prevalence of concomitant auto-immune rheumatic disease in IIM compared to the SLE and Sjogren's population.
References
Ehrenstein MR, Snaith ML, Isenberg DA (1992) Idiopathic myositis: a rheumatological view. Ann Rheum Dis 51(1):41–44
Sultan SM, Ioannou Y, Moss K, Isenberg DA (2002) Outcome in patients with idiopathic inflammatory myositis: morbidity and mortality. Rheumatology (Oxford) 41(1):22–26
Marie I, Hachulla E, Hatron PY et al (2001) Polymyositis and dermatomyositis: short term and long term outcome, and predictive factors of prognosis. J Rheumatol 28(10):2230–2237
Ponyi A, Borgulya G, Constantin T, Vancsa A, Gergely L, Danko K (2005) Functional outcome and quality of life in adult patients with idiopathic inflammatory myositis. Rheumatology (Oxford) 44(1):83–88
Airio A, Kautiainen H, Hakala M (2006) Prognosis and mortality of polymyositis and dermatomyositis patients. Clin Rheumatol 25(2):234–239
Torres C, Belmonte R, Carmona L et al (2006) Survival, mortality and causes of death in inflammatory myopathies. Autoimmunity 39(3):205–215
Maugars YM, Berthelot JM, Abbas AA, Mussini JM, Nguyen JM, Prost AM (1996) Long-term prognosis of 69 patients with dermatomyositis or polymyositis. Clin Exp Rheumatol 14(3):263–274
Tymms KE, Webb J (1985) Dermatopolymyositis and other connective tissue diseases: a review of 105 cases. J Rheumatol 12(6):1140–1148
Rios G (2005) Retrospective review of the clinical manifestations and outcomes in Puerto Ricans with idiopathic inflammatory myopathies. J Clin Rheumatol 11(3):153–156
Uthman I, Vázquez-Abad D, Senécal JL (1996) Distinctive features of idiopathic inflammatory myopathies in French Canadians. Semin Arthritis Rheum 26(1):447–458
McDonagh JE, Isenberg DA (2000) Development of additional autoimmune diseases in a population of patients with systemic lupus erythematosus. Ann Rheum Dis 59(3):230–232
Chambers SA, Charman SC, Rahman A, Isenberg DA (2007) Development of additional autoimmune diseases in a multiethnic cohort of patients with systemic lupus erythematosus with reference to damage and mortality. Ann Rheum Dis 66(9):1173–1177
Lazarus MN, Isenberg DA (2005) Development of additional autoimmune diseases in a population of patients with primary Sjogren's syndrome. Ann Rheum Dis 64(7):1062–1064
Bohan A, Peter JB (1975) Polymyositis and dermatomyositis (first of two parts). N Engl J Med 292(7):344–347
Bohan A, Peter JB (1975) Polymyositis and dermatomyositis (second of two parts). N Engl J Med 292(8):403–407
Vitali C, Bombardieri S, Moutsopoulos HM et al (1993) Preliminary criteria for the classification of Sjögren's syndrome. Results of a prospective concerted action supported by the European Community. Arthritis Rheum 36(3):340–347
Clarke AE, Bloch DA, Medsger TA Jr, Oddis CV (1995) A longitudinal study of functional disability in a national cohort of patients with polymyositis/dermatomyositis. Arthritis Rheum 38(9):1218–1224
Bronner IM, van der Meulen MF, de Visser M et al (2006) Long-term outcome in polymyositis and dermatomyositis. Ann Rheum Dis 65(11):1456–1461
Marie I, Hachulla E, Cherin P et al (2002) Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 47(6):614–622
Selva-O'Callaghan A, Labrador-Horrillo M, Munoz-Gall X et al (2005) Polymyositis/dermatomyositis-associated lung disease: analysis of a series of 81 patients. Lupus 14(7):534–542
Yaqub S, Moder KG, Lacy MQ (2004) Severe, reversible pulmonary hypertension in a patient with monoclonal gammopathy and features of dermatomyositis. Mayo Clin Proc 79(5):687–689
Grateau G, Roux ME, Franck N et al (1993) Pulmonary hypertension in a case of dermatomyositis. J Rheumatol 20(8):1452–1453
Kissel JT, Mendell JR, Rammohan KW (1986) Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med 314(6):329–334
Wakata N, Kurihara T, Saito E, Kinoshita M (2002) Polymyositis and dermatomyositis associated with malignancy: a 30-year retrospective study. Int J Dermatol 41(11):729–734
Hill CL, Zhang Y, Sigurgeirsson B et al (2001) Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet 357(9250):96–100
Sigurgeirsson B, Lindelof B, Edhag O, Allander E (1992) Risk of cancer in patients with dermatomyositis or polymyositis. A population-based study. N Engl J Med 326(6):363–367
Bernatsky S, Boivin JF, Joseph L et al (2005) An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 52(5):1481–1490
Bernatsky S, Clarke AE, Suissa S (2008) Haematologic malignant neoplasms after drug exposure in rheumatoid arthritis. Arch Intern Med 168(4):378–381
Biro E, Szekanecz Z, Czirjak L et al (2006) Association of systemic and thyroid autoimmune diseases. Clin Rheumatol 25(2):240–245
Koh ET, Seow A, Ong B, Ratnagopal P, Tjia H, Chng HH (1993) Adult onset polymyositis/dermatomyositis: clinical and laboratory features and treatment response in 75 patients. Ann Rheum Dis 52(12):857–861
Shamim EA, Miller FW (2000) Familial autoimmunity and the idiopathic inflammatory myopathies. Curr Rheumatol Rep 2(3):201–211
Marie I, Hachulla E, Cherin P et al (2005) Opportunistic infections in polymyositis and dermatomyositis. Arthritis Rheum 53(2):155–165
Viguier M, Fouere S, de la Salmoniere P et al (2003) Peripheral blood lymphocyte subset counts in patients with dermatomyositis: clinical correlations and changes following therapy. Medicine (Baltimore) 82(2):82–86
Ishida Y, Utikoshi M, Kurosaki M et al (1998) Hepatic veno-occlusive disease in a case of polymyositis associated with thrombotic thrombocytopenic purpura/hemolytic uremic syndrome. Intern Med 37(8):694–699
Miller FW, Rider LG, Chung YL et al (2001) International Myositis Outcome Assessment Collaborative Study Group. Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford) 40(11):1262–1273
Isenberg DA, Allen E, Farewell V et al (2004) International Myositis and Clinical Studies Group (IMACS). International consensus outcome measures for patients with idiopathic inflammatory myopathies. Development and initial validation of myositis activity and damage indices in patients with adult onset disease. Rheumatology (Oxford) 43(1):49–54
Sultan SM, Allen E, Oddis CV et al (2008) Reliability and validity of the myositis disease activity assessment tool. Arthritis Rheum 58(11):3593–3599
Shamim EA, Rider LG, Pandey JP et al (2002) Differences in idiopathic inflammatory myopathy phenotypes and genotypes between Mesoamerican mestizos and North American Caucasians: ethnogeographic influences in the genetics and clinical expression of myositis. Arthritis Rheum 46(7):1885–1893
O'Hanlon TP, Rider LG, Mamyrova G et al (2006) HLA polymorphisms in African Americans with idiopathic inflammatory myopathy: allelic profiles distinguish patients with different clinical phenotypes and myositis autoantibodies. Arthritis Rheum 54(11):3670–3681
Acknowledgements
Dr. Kristine P. Ng is a recipient of the New Zealand Rose Hellaby medical scholarship.
Disclosures
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ng, K.P., Ramos, F., Sultan, S.M. et al. Concomitant diseases in a cohort of patients with idiopathic myositis during long-term follow-up. Clin Rheumatol 28, 947–953 (2009). https://doi.org/10.1007/s10067-009-1181-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-009-1181-4