
Abstract
Systemic lupus erythematosus (SLE) is the prototype autoimmune disorder, one that is known for its many, diverse modes of presentation. In this paper, we present a further unusual presentation of SLE, that of acute onset, severe heart failure secondary to dilated cardiomyopathy. Only a few similar cases have been reported in the literature.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Case
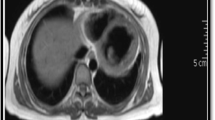
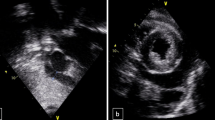
A 32-year-old Caucasian woman was admitted to our hospital because of palpitations, malaise, weakness, severe dyspnea, and orthopnea over the preceding 10 days. Her previous medical history was unremarkable. She had no family history of relevant illness. On initial evaluation, she was in a rather poor clinical condition and was confused. Blood pressure was 110/80 mmHg, pulse rate was 124 beats/min, respiratory rate was 32/min, and body temperature was 38.7°C. Physical examination revealed cool skin, peripheral cyanosis, tachypnea, tachycardia, jugular venous distension, pulsus alternans, a third heart sound, crackles over the middle and basal lung areas, and hepatomegaly. Chest x-ray disclosed symmetrical globular enlargement of the heart and pulmonary congestion in a perihilar distribution, with a small pleural effusion in the left sinus (Fig. 1). An electrocardiogram revealed sinus tachycardia with ST-T wave abnormalities. Echocardiogram demonstrated dilatation of all cardiac chambers with severe, global contractile impairment and a 24% ejection fraction. Severe mitral and tricuspid regurgitation was observed on color-flow Doppler echocardiographic examination. Coronary angiography was normal. Initial laboratory studies revealed a hemoglobin level of 10.2 g/dl with a normochromic, normocytic peripheral smear. White blood cell count was 10,200 with 69% polymorphs, 23% lymphocytes, 6% monocytes, and 2% eosinophils. Platelet count was 252,000/mm3. Erythrocyte sedimentation rate was 90 mm/h. Serum urea nitrogen and creatinine levels were 28 and 1.0 mg/dl, respectively. Electrolyte concentrations were normal. Serum transaminases were mildly elevated with 62 U/l alanine transaminase and 72 U/l aspartate transaminase. Hematologic studies were compatible with findings of hemolysis [high serum lactate dehydrogenase (720 U/l; normal range 230–480 U/l), high total/indirect bilirubin levels (2.4/1.8 mg/dl; normal range <1.6/<1.0 mg/dl), increased reticulocyte count (3.5%; normal range 0.8–1.0%), and reduced serum levels of haptoglobins (12 mg/dl; normal range 40–230 mg/dl) with a positive direct Coombs’ test]. The results of urinalysis were specific gravity 1.016, pH 6.5, with +2 protein, and urine sediment examination revealed a small number of erythrocytes. She had 780 mg protein in a 24-h urine collection. Serological examinations for viral infections, including herpes simplex, cytomegalovirus, Epstein-Barr, coxsackievirus, enterovirus, rubella, rubeola, influenza, hepatitis B and C, and HIV, were all negative. Thyroid hormone levels were within normal limits. Serological tests indicated CH50 24.8 U/ml (normal 30–40), C3 28.4 mg/dl (normal 40–130), and C4 8.2 mg/dl (normal 20–40); antinuclear antibody (ANA) with a speckled pattern was detected at a dilution of 1:160; anti-dsDNA antibody titer was 98.6 U/ml (normal 0–7), and an anti-cardiolipin immunoglobulin M (IgM) test was positive. All other immunologic markers, including anti-Ro, anti-La, anti-Sm, anti-ribonucleoprotein, anti-cardiolipin IgG, LE cells, VDRL, and RF, were negative.

Chest x-ray showing a symmetrical globular enlargement of the heart with pulmonary congestion in perihilar distribution
In accordance with the above-mentioned findings, a diagnosis of dilated cardiomyopathy leading to heart failure was made. Furosemide, an angiotensin-converting enzyme (ACE) inhibitor, and digoxin were intravenously administered immediately. However, her overall condition did not improve, despite maximal inotropic support. Since the patient’s clinical and laboratory presentation was compatible with systemic lupus erythematosus (SLE), 80 mg daily of prednisolone was added to treatment. The patient’s clinical condition improved within 10 days, and 1 month from initiation of therapy, a repeat chest x-ray showed marked diminution of cardiac size (Fig. 2). Echocardiography was repeated and revealed improvement in global contractile function to a 55% ejection fraction and diminution in the size of all cardiac chambers. Consequently, prednisolone dose was gradually tapered up to 10 mg daily over a 2-month period. Three months after initiation of therapy, global general improvement was observed in the patient’s clinical picture. Chest x-ray and echocardiographic examinations revealed complete resolution of the previous abnormal findings. Her immunologic status had improved as well (anti-dsDNA titers reduced to 12 IU/ml, and serum complements levels returned to their normal range). No laboratory findings suggesting hemolysis or renal disorder persisted. Therapy was maintained with only a low-dose ACE inhibitor and prednisolone (10 mg on alternate days).

Control chest x-ray showing marked diminutation of cardiac size
Discussion
This patient posed several clinical dilemmas. First, what was the cause of her heart failure? This woman’s age and clinical presentation made coronary artery disease, valvular heart disease, and hypertensive cardiomyopathy all highly unlikely. She presented with severe and rapid-onset heart failure, global cardiac enlargement on chest x-ray, and cardiac chamber dilatation and global contractile impairment on echocardiogram but with no prior history of hypertension or other significant illness and a normal coronary angiography. Hence, dilated cardiomyopathy seemed the most probable explanation. This realization, however, brought us to our second dilemma. What was the etiology of her dilated cardiomyopathy?
Dilated cardiomyopathy may occur idiopathically or in association with a variety of clinical settings, including viral illness, alcoholism, doxorubicin toxicity, muscular dystrophy, pregnancy or the postpartum period, and the autoimmune collagen vascular diseases [1]. In addition, thyrotoxicosis rarely may present as a dilated cardiomyopathy. There is a rare form of hereditary X-linked dilated cardiomyopathy. In this patient, most of these possibilities could be ruled out by clinical history. Thyrotoxicosis was eliminated as a possibility when thyroid screening tests returned were within normal range. This reduced the list of possible etiologies to viral, autoimmune, and idiopathic cardiomyopathy.
Although occult viral infection likely is an underlying cause in many of the so-called “idiopathic” cases [2], the immunologic picture in this case was not consistent with this. The hemolytic anemia, proteinuria, pleural effusion, and positive ANA and anti-dsDNA titers seemed most consistent with a diagnosis of autoimmune collagen vascular disease and, in particular, with SLE. This suggested a possible link between dilated cardiomyopathy and SLE. Indeed, we could not identify any etiologic possibility other than SLE to explain this patient’s presentation.
SLE is an inflammatory, autoimmune disease of unknown etiology, characterized by the production of autoantibodies and the deposition of immune complexes in various organs. It has the potential to affect virtually all organ systems [3]. It can be difficult to differentiate from other connective tissue disorders in its early stages. For this reason, the American College of Rheumatology (formerly the American Rheumatism Association) developed diagnostic criteria for SLE, which include four different mucocutaneous manifestations (malar rash, photosensitivity rash, discoid lesions, and mucous ulcers), inflammatory arthritis, serositis (either pleuritis or pericarditis), neurological involvement, renal involvement, hematologic involvement (anemia, leukopenia, thrombocytopenia, or pancytopenia), positive ANA, and specific antinuclear antibodies (anti-dsDNA or anti-Sm). Any 4 of these 11 criteria are said to establish a diagnosis of definite SLE [3]. Cardiovascular involvement occurs frequently, although it is often mild enough not to cause clinical concern [4]. Pericarditis is the most frequent form of cardiac involvement. Although lupus percarditis most commonly is subclinical, noted only on echocardiogram, it may be associated with clinical symptoms and signs of pericardial effusion and uncommonly requires pericardial aspiration or surgical preparation of a pericardial window. Rarely, tamponade or late obstruction can occur. Valvular heart diseases related to Libman–Sacks endocarditis are also rare cardiovascular manifestations of SLE. These valvular lesions may embolize or become infected. The frequency of ischemic heart disease secondary to premature atherosclerosis or coronary arteritis is increased in SLE [4, 5]. Although evidence of myocarditis frequently has been observed in series of autopsies of patients with SLE, the incidence and prevalence of clinically apparent primary myocardial dysfunction appears to be low [6]. Myocardial involvement usually accompanies other cardiac lesions. Isolated myocarditis, or dilated cardiomyopathy, is a rare and usually late clinical manifestation of SLE. Late cardiomyopathy associated with SLE may be due to thrombotic or inflammatory microvascular coronary disease [6].
Dilated cardiomyopathy is a disorder associated with myocardial dysfunction that is predominantly related to ventricular dilatation. Alterations in ventricle size and shape lead to secondary functional mitral and/or tricuspid regurgitation and atrial dilatation. The physiological consequence of this pathological process is primarily depression of ventricular systolic function reflected by a low election fraction [1]. Management of these patients includes treatment of heart failure, adjuvant treatment strategies (anticoagulation, antiarrhythmic drugs), treatment or correction of the underlying cause (if possible), and cardiac transplantation. Steroids and/or other immunosupressive drugs may be effective for autoimmune cardiomyopathy. Anecdotally, patients generally seem to respond well and rapidly to these agents. There is no indication for standard use of steroids in dilated cardiomyopathy other than when the underlying process is felt to be autoimmune [7].
This brings us to the third and final dilemma with our patient: how should we treat her? Because we felt confident that her cardiomyopathy was related to previously undiagnosed SLE, we also felt that a course of steroids was warranted, in conjunction with supportive therapy using a diuretic, an ACE inhibitor, and digoxin. Ultimately, our intuition was correct, because her clinical condition improved dramatically, her ejection fraction approaching normal within 3 months after initiation of therapy.
This patient, admitted with severe heart failure secondary related to dilated cardiomyopathy as the initial clinical presentation of SLE, is of interest because of the rarity of this presentation; to our knowledge, only a few similar cases have been reported before [8, 9]. It was not until we initiated clinical investigations that any classic clinical manifestations of SLE were detected. On the other hand, it was not until we initiated corticosteroid therapy, in addition to traditional management for heart failure, that her clinical picture improved. This leaves little doubt that the diagnosis of autoimmune cardiomyopathy secondary to SLE was correct.
In conclusion, dilated cardiomyopathy is a rare initial clinical manifestation of SLE. Diagnosis can be difficult if there are no classical findings of SLE. Nonetheless, the possibility of cardiomyopathy secondary to SLE must always be considered in the differential diagnosis of heart failure of unknown etiology that is unresponsive to maximal inotropic support and high-dose diuretic therapy, especially in a young woman. Although the risks of chronic corticosteroid use cannot be overstated, corticosteroids alone or, possibly, in combination with other immunosuppressive drugs may be an effective treatment for this clinical condition.
Refrences
Kasper EK, Agema WRP, Hutchins GM, Deckers JW, Hare JM, Baughman KL (1994) The causes of dilated cardiomyopathy: a clinicopathologic review of 673 consecutive patients. J Am Coll Cardiol 23:586–590
Bender JR (1991) Idiopathic dilated cardiomyopathy: an immunologic, genetic, or infectious disease, or all of the above? Circulation 83:704–710
The American Rheumatism Association (1982) The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25:1271
Mandell BF (1987) Cardiovascular involvement in systemic lupus erythematosus. Semin Arthritis Rheum 17(2):126–141
Badui E, Garcia-Rubi D, Robles E, Jimenez J, Juan L, Deleze M, Diaz A, Mintz G (1985) Cardiovascular manifestations in systemic lupus erythematosus. Prospective study of 100 patients. Angiology 36(7):431–441
Murai K, Oku H, Takeuchi K, Kanayama Y, Inoue T, Takeda T (1987) Alterations in myocardial systolic and diastolic function in patients with active systemic lupus erythematosus. Am Heart J 113:966–971
Parrillo JE, Cunnion RE, Epstein SE, Parker MM, Sufredini AF, Brenner M, Schaer GL, Palmeri ST, Cannon RO, Alling D, Wittes JT, Ferrans VJ, Rodriguez ER, Fauci AS (1989) A prospective, randomized, controlled trial of prednisone for dilated cardiomyopathy. N Engl J Med 321:1061–1068
Sandrasegaran K, Clarke CW, Nagendran V (1992) Sub-clinical systemic lupus erythematosus presenting with acute myocarditis. Postgrad Med J 68(800):475–478
Frustaci A, Gentiloni N, Caldarulo M (1996) Acute myocarditis and left ventricular aneurysm as presentations of systemic lupus erythematosus. Chest 109(1):282–284
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Usalan, C., Buyukhatipoglu, H. & Tiryaki, O. Systemic lupus erythematosus complicated by dilated cardiomyopathy and severe heart failure. Clin Rheumatol 26, 125–127 (2007). https://doi.org/10.1007/s10067-005-0123-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-005-0123-z