
ABSTRACT
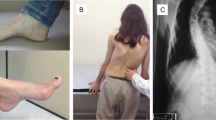
Most cases of Friedreich ataxia (FRDA) are due to expansions of a GAA trinucleotide repeat sequence in the FRDA gene coding for frataxin, a protein of poorly understood function which may regulate mitochondrial iron transport. However, between 1% and 5% of mutations are single base changes in the sequence of the FRDA gene, causing missense, nonsense, or splicing mutations. We describe three new mutations, IVS4nt2 (T to G), R165C , and L182F , which occur in patients in association with GAA expansions. These cases, and a further five reported cases of point mutations causing FRDA, demonstrate that splicing, nonsense, or initiation codon mutations (which cause a complete absence of functional frataxin) are associated with a severe phenotype. Missense mutations, even in highly evolutionally conserved amino acids, may cause a mild or severe phenotype.
Article PDF
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Author information
Authors and Affiliations
Additional information
Received: March 24, 1998 / Accepted: May 28, 1998
Rights and permissions
About this article
Cite this article
Forrest, S., Knight, M., Delatycki, M. et al. The correlation of clinical phenotype in Friedreich ataxia with the site of point mutations in the FRDA gene. Neurogenetics 1, 253–257 (1998). https://doi.org/10.1007/s100480050037
Issue Date:
DOI: https://doi.org/10.1007/s100480050037