
Abstract
Dandy-Walker malformation (DWM) has been reported to have heterogeneous causes, including mutations in genes of fibroblast growth factors and in genes in the sonic hedgehog (Shh) signaling pathway. Here, we identified an activating cancerous inhibitor of protein phosphatase 2A (CIP2A) p.D269V mutation, located at the predicted protein-protein interaction groove, as a novel genetic cause of Dandy-Walker variant (DWV). CIP2A has been reported as an oncoprotein promoting tumor survival via inhibition of protein phosphatase 2A (PP2A). However, the impact of human germline CIP2A mutation is unknown. We report a novel heterozygous CIP2A p.D269V mutation via whole exome sequencing in two siblings with DWV and severe intellectual disability who were born to non-consanguineous parents. Only the older brother developed a slow-growing sacral leiomyoma in his teens. The CIP2A p.D269V mutation is associated with increased PP2A, mTOR, and c-Myc protein levels in peripheral blood mononuclear cells (PBMCs). The PP2A phosphatase activity, however, was not suppressed. Deep sequencing revealed that the father carries 16% of somatic CIP2A p.D269V mutation, suggesting potential inheritance from the mosaic sperm populations. Our study is the first to describe a pathogenic CIP2A mutation in humans, which might disrupt neuronal development via enhancing mTOR and c-Myc protein expressions, shedding light in mechanisms of DWV pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dandy-Walker malformation (DWM) primarily affects the development of the cerebellum, resulting in partial or complete agenesis of the cerebellar vermis, and cystic dilatation of the fourth ventricle, with or without hydrocephalus (OMIM 220200). For the terminology, DWM is at one end of a spectrum of posterior fossa anomalies (Dandy-Walker complex) in children, with enlargement of posterior fossa and elevation of the confluence of sinuses, while those cases with similar neuroradiological images but with a normal-sized posterior fossa fall in the category of Dandy-Walker variant (DWV) [1]. The etiology of DWM/DWV is heterogeneous. Mutations in several genes of the sonic hedgehog (Shh) signaling pathway, including ZIC1 and ZIC4, have been reported to be causes of subsets of DWM patients [2, 3]. Mutations in fibroblast growth factor genes FGF8, FGF17, and in other genes such as LAMC1, FOXC1, and FOXL2, have also been implicated in DWM [4,5,6,7]. Furthermore, both FGF8 and WNT1 signaling molecules are known to be important in neural tube patterning and cerebellar development [8]. Deletion in FGF8 has been reported to be associated with vermis hypoplasia, one of the features in DWV [9]. Due to its low empiric recurrence risk, it is suggested that mendelian inheritance is unlikely for non-syndromic DWM [10].
Cancerous inhibitor of protein phosphatase 2A (CIP2A) has been reported as an oncoprotein promoting tumor survival via inhibition of protein phosphatase 2A (PP2A) causing AKT activation or MYC stabilization [11]. CIP2A can also interact with the mTOR pathway, which might influence neuronal development [12]. It has been reported that mutations in the PI3K-AKT-mTOR pathway could cause brain overgrowth syndrome and neuronal migration defects [3]. The impact of gain-of-function CIP2A germline mutation on human diseases is currently unclear. Here, we present two siblings with DWV, developmental delay, and intellectual disability, who are carriers of an activating CIP2A p.D269V mutation causing enhanced PP2A, mTOR, and c-Myc protein expressions. Deep sequencing of CIP2A in the parents revealed potential inheritance from the mosaic sperm populations.
Materials and methods
Patients
We studied a non-consanguineous family of two affected children with DWM and severe intellectual disability. The study was approved by the Research Ethics Committee of China Medical University Hospital (CMUH106-REC1-047). Written informed consents were obtained from the participants.
Whole exome sequencing and data analysis
Peripheral blood mononuclear cells (PBMCs) were collected from the index case and DNA, RNA, and proteins were extracted via standard procedures for further analyses. DNA libraries were prepared using Illumina Truseq exome Library Prep kit, and sequenced on the Illumina NextSeq 500 platform. Base calling and quality scoring were performed by an updated implementation of real-time analysis (RTA) on NextSeq 500. Bcl2fastq Conversion Software was used to demultiplex data and to convert BCL files to FASTQ file formats. Sequenced reads were trimmed for low-quality sequence, then aligned to the human reference genome (hg19) using BWA [13]. Finally, SNPs and small INDELs were called in individual samples by GATK [14] and VarScan [15] at their default settings. We then performed ANNOVAR [16] to functionally annotate genetic variants. The following criteria were used to select potential candidates: the mutant allele frequency ≥ 30%, global minor allele frequency < 1%, and exclusion of variants showing “benign” clinical significance in ClinVar, predicted to be pathogenic by at least one of the three software: SIFT, PolyPhen, and CADD_PHRED. Fifty-three genes related to DWM reported in the literatures [2, 4,5,6,7, 17] and genes recommended by ACMG for reporting incidental findings [18] were annotated. After the identification of potentially causative variants, confirmation was performed by Sanger sequencing.
Protein quantification and phosphatase activity assay
Western blot analysis was performed to detect the PBMC protein expressions using antibodies against CIP2A (SANTA, #2G10-3B5), PP2A (Millipore, #2880678), mTOR (St John’s Laboratory, #STJ94280), c-Myc (SANTA, #C-33), and actin (Abcam, #ab8226, as internal control). The CIP2A serum levels were detected by ELISA kit (MyBioSource). PP2A phosphatase activity assay was performed by using PP2A immunoprecipitation phosphatase assay kit (Merk, Millipore).
Lymphocyte activation assay
PBMCs were rested overnight and the suspended lymphocytes were added to the anti-CD3-coated (eBioscience, #4310640, 10 μg/ml) wells together with anti-CD28 (eBioscience, #4311047, 10 μg/ml). RNAs were extracted from the stimulated and the non-stimulated cells after 6 h via TRIzol reagent and reverse-transcribed to cDNA. Real-time PCR was performed to detect the expression levels of IFN-γand IL-2 using the respective primers.
Statistical analysis
The non-parametric Mann-Whitney U tests using GraphPad Prism 5 were applied to compare PBMC protein expression levels between patients and healthy family members. String Version 10.5 was used to analyze potential interactions among CIP2A, PP2A, mTOR, Myc, and molecules related to DWM or cerebellar development.
Results
Clinical features and brain MRI studies
The index case is a 24-year-old male, who was born to healthy non-consanguineous parents at full term. The prenatal ultrasonography 24 years ago did not detect brain abnormality, but small head circumference was noted at gestational age of 34 weeks. He had neonatal hypotonia and could be pulled to sit with head lag at around 8.5-month-old. He suffered from unsteady gait, together with severe intellectual disability and autistic behaviors. He had difficulty in learning and was poorly oriented. Furthermore, at 24-year-old, he could only make sounds, neither speech nor language was developed. When growing up, there was no seizure episode or frequent infections. On physical examination, his adult head circumference was within normal limits, but high forehead with normally positioned and configured ear auricles were noted. There was a soft, slow-growing mass at his sacral area, which was first noticed at age 14. Ultrasound examinations of heart, kidneys, and abdomen showed no anomalies. Previous chromosomal analysis showed a normal male 46, XY karyotype. The patient has an apparently normal younger sister and an affected younger sister (18-year-old) who presented with similar clinical phenotypes since early childhood, including unsteady gait, developmental delay, and severe intellectual disability. As compared with the index case, the affected younger sister did not have palpable mass lesion and had amblyopia in addition to the clinical presentations described above.
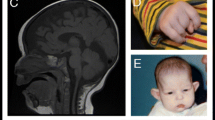
The brain MRI of both patients revealed hypoplasia of vermis especially at the inferior parts, and enlargement of the fourth ventricle along the cystic space of the posterior fossa (Fig. 1a, b). However, the posterior fossa was not enlarged and the confluence of sinuses (torcula) was not elevated. Neither supratentorial hydrocephalus nor atrophy of the bilateral cerebral hemispheres was observed. These findings suggest the diagnosis of Dandy-Walker variant (DWV). Furthermore, significant atrophy of the pons and mild atrophy of the inferior part of the midbrain were detected. The bilateral cerebellar hemispheres with apparently normal architecture were reduced to a very small volume, which could be found in the brain MRI (Fig. not shown). Pons and midbrain atrophies might be secondary to the lack of interaction with the bilateral cerebellar hemispheres. Sagittal view of T1-weighted FLAIR image of the affected sister (S(Pt)) at age 18 showed the same findings (Fig. 1b) as the index case (Fig. 1a). In addition, spinal MRI of the older brother showed a sacral leiomyoma, which grew to about 4 cm in the longest diameter over 10 years (Fig. 1c, d).

Neuroimaging studies of the affected siblings. a Sagittal view of T1-weighted flair image of the index case (Pt) at age 24, showing hypoplasia of vermis especially at the inferior parts, and enlargement of the fourth ventricle along the cystic space of the posterior fossa. The posterior fossa was not enlarged and the confluence of sinuses (torcula) was not elevated. Neither supratentorial hydrocephalus nor atrophy of the bilateral cerebral hemispheres was observed, but significant atrophy of the pons and mild atrophy of the inferior part of the midbrain were detected. These findings suggest the diagnosis of Dandy-Walker variant. b Sagittal view of T1-weighted flair image of the affected sister (S(Pt)) at age 18, showing similar findings as the index case. c, d The appearance (c) and axial CT with contrast image (d) of the sacral leiomyoma of the index case at age 24
CIP2A c.806A>T (p.D269V) mutation identified as the genetic cause
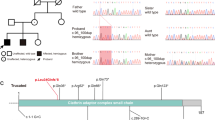
Our whole exome analysis pipeline found 6 candidate variants (ZNF107 p.I737fs, PSEN2 p.T142fs, SHISA8 p.265_270del, PIK3CB p.R493W, CERCAM p.R402C, CIP2A p.D269V) in the index case. Further, Sanger sequencing of the above genes in PBMCs of the family members (parents, unaffected sister, and affected sister) showed that CIP2A p.D269V is the unique mutation seen only in the two affected siblings. To increase the sensitivity of mutation detection, deep sequencing of the family DNAs for the CIP2A variant was performed via Illumina NextSeq 500, which revealed the CIP2A p.D269V mutation load to be 50, 60 and 16% in the index case, the affected sister, and the father, respectively (Fig. 2).

Genetic analyses. Pedigree with percentages of the CIP2A p.D269V mutation in PBMCs of the proband and the family members, determined by next-generation sequencing
Functional impacts of the CIP2A c.806A>T (p.D269V) mutation
Since CIP2A p.D269V is located at the predicted protein-protein interaction area [19] and has been reported to interact with PP2A, c-Myc, and mTOR [11], we performed western blot analysis for the aforementioned proteins in PBMC lysates derived from the index case and his family members (Fig. 3a). The cell lysate CIP2A expression varied among individuals, and as compared with the unaffected family members, only a mild elevation of serum CIP2A level was detected in both affected patients (Figs. 3a, b and 4a). In contrast, the mTOR protein levels in the two affected siblings were around twofold as compared with samples of other unaffected individuals, and the PP2A protein levels were also higher in the patients (Fig. 3a, c, and d). The c-Myc protein expressions were mildly elevated in the probands (Fig. 3a). The PP2A phosphatase activities were not suppressed in both cases with DWV (Fig. 4b). Further, network analysis of CIP2A, PP2A, mTOR, Myc, and molecules related to DWM or cerebellar development revealed potential interactions among mTOR, Myc, Shh, and Wnt-1 (Fig. 5).

PBMC protein expression levels among patients and unaffected family members. a Western blot analysis of relative PBMC protein expression levels as compared with actin. Pt, the affected brother; F, father; M, mother; S(N), the unaffected sister; S(Pt), the affected sister. The numbers shown in each lane represent the expressions as a ratio with actin. This is a representative blot picture of multiple experiments (b, c). b PBMC CIP2A protein expression levels of patients and family members among different experiments. The protein levels of the mother were evaluated in all experiments and were used as an inter-experimental control. Actin was the intra-experimental control. Protein expression levels were first compared with actin and shown as fold of the mother. Lines represent medians; Mann-Whitney U test was performed to analyze the difference of CIP2A protein expression levels between patients (Pt, S(Pt)) and apparently normal family members (F, M, S(N)). ns, not statistically significant. c PBMC mTOR protein expression levels of patients and family members among different experiments. *p < 0.05 by Mann-Whitney U test. d PBMC PP2A protein expression levels of patients and family members among different experiments. ***p < 0.001 by Mann-Whitney U test

Functional studies of the CIP2A mutation. a Serum CIP2A protein levels (n = 6, bars represent mean ± SEM). b PBMC PP2A phosphatase activities of the patients and family members. Pt, the affected brother; F, father; M, mother; S(N), the unaffected sister; S(Pt), the affected sister

Network analysis of CIP2A, PP2A, mTOR, Myc, and molecules involving neural tube patterning and cerebellar development revealed potential interactions among mTOR, Myc, Shh, and Wnt-1. The analysis was performed using String Version 10.5. Light green lines connecting different molecules represent potential interactions (the two molecules have been co-mentioned in Pubmed abstracts). Enriched biological processes as analyzed by String Version 10.5 are listed below the graph
Effects of CIP2A c.806A>T (p.D269V) on lymphocyte activation
Lymphocyte activation assay revealed that as compared with the unaffected family members, the affected siblings had impaired T cell IFN-γand IL-2 responses upon CD3 and CD28 stimulations (Fig. 6a, b).

Activation assays of lymphocytes derived from patients and family members (a, b). Lymphocytes were activated with anti-CD3 and anti-CD28 for 6 h, and the IFN-γ(a) and IL-2 (b) mRNA levels were measured as compared with baseline levels in unstimulated lymphocytes
Discussion
We described here CIP2A p.D269V mutation as potential genetic cause of DWV and intellectual disability seen in two siblings born to non-consanguineous parents. Interestingly, the asymptomatic father also carried 16% of the CIP2A p.D269V mutation, suggesting inheritance from the mosaic sperm populations.
The human CIP2A gene sequence is conserved in mice, and CIP2A protein is reported to be expressed at a moderate amount in mouse embryonic brain (OMIM 610643). According to the Human Protein Atlas (https://www.proteinatlas.org), CIP2A is highly expressed in the brain, especially at the cerebellum. Although CIP2A mutations have been frequently found in different types of human cancers (COSMIC database), CIP2A p.D269V mutation has never been reported, and we are the first to demonstrate a potential correlation of this variant with human cerebellar malformation and intellectual disability.
As reviewed by Pradip De et al., CIP2A has multiple functions, including PP2A inactivation, c-Myc stabilization, and mTORC1 activation [11]. The 3D-structure of CIP2A was predicted to be a stable armadillo repeat fold with a positively charged groove, and the CIP2A p.D269V mutation site is approximately at the groove, which potentially involves peptide binding [19]. Besides their roles in tumor progression, c-Myc has been demonstrated to influence neural precursor cell fate and direct cerebellar development [20], while gain-of-function mutations in mTOR have been reported to cause megalencephaly and other malformations in brain development [12]. Functional studies on PBMCs with CIP2A p.D269V mutation revealed mildly elevated serum CIP2A level, and increased protein expressions of PP2A, mTOR, and c-Myc. Network analysis of CIP2A, PP2A, mTOR, Myc, and molecules involving neural tube patterning and cerebellar development revealed potential interactions among mTOR, Myc, Shh, and Wnt-1. Therefore, it is possible that CIP2A p.D269V contributed to DWV and intellectual disability via enhanced mTOR and c-Myc activities. Germline genome editing is needed in animal models to validate the causal relationship between CIP2A p.D269V and cerebellar malformation.
In addition to its expression in the brain, CIP2A is reported to be expressed at significant levels at the testis and lymph nodes in both human and mice (OMIM 610643, https://www.proteinatlas.org). Hypomorphic CIP2AHOZ mice had been generated and demonstrated defects in spermatogenesis and T cell activations [21, 22]. However, mice with activating CIP2A mutations have not been generated. We observed increased mTOR expression in resting CIP2A p.D269V lymphocytes. MTOR is a key molecule integrating the environmental cues determining T cell differentiation and proliferation [23]. The impaired IFN-γand IL-2 response to TCR stimulations seen in CIP2A p.D269V lymphocytes could be an indirect consequence of an already activated mTOR signaling at baseline. Phenotypically, the probands showed no history of recurrent infections and no malignant tumors were detected over 20 years, suggesting that their immunity is generally intact. The affected siblings are regularly followed up at the neurology clinic for check-ups of potential development of malignancy.
Conclusion
Our study is the first to describe an activating pathogenic CIP2A genetic variant in humans, which is inherited from paternal post-zygotic somatic mutation. The CIP2A p.D269V mutation might disrupt neuronal development via enhancing mTOR and c-Myc protein expressions, shedding light on novel mechanisms of DWV pathogenesis.
References
Sasaki-Adams D, Elbabaa SK, Jewells V, Carter L, Campbell JW, Ritter AM (2008) The Dandy-Walker variant: a case series of 24 pediatric patients and evaluation of associated anomalies, incidence of hydrocephalus, and developmental outcomes. J Neurosurg Pediatr 2(3):194–199
Blank MC, Grinberg I, Aryee E, Laliberte C, Chizhikov VV, Henkelman RM, Millen KJ (2011) Multiple developmental programs are altered by loss of Zic1 and Zic4 to cause Dandy-Walker malformation cerebellar pathogenesis. Development 138(6):1207–1216
Dyment DA, Sawyer SL, Chardon JW, Boycott KM (2013) Recent advances in the genetic etiology of brain malformations. Curr Neurol Neurosci Rep 13(8):364
Zanni G, Barresi S, Travaglini L, Bernardini L, Rizza T, Digilio MC, Mercuri E, Cianfarani S, Valeriani M, Ferraris A, Da Sacco L, Novelli A, Valente EM, Dallapiccola B, Bertini ES (2011) FGF17, a gene involved in cerebellar development, is downregulated in a patient with Dandy-Walker malformation carrying a de novo 8p deletion. Neurogenetics 12(3):241–245
Aldinger KA, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC, Krantz ID, Dobyns WB, Millen KJ (2009) FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet 41(9):1037–1042
Lim BC, Park WY, Seo EJ, Kim KJ, Hwang YS, Chae JH (2011) De novo interstitial deletion of 3q22.3-q25.2 encompassing FOXL2, ATR, ZIC1, and ZIC4 in a patient with blepharophimosis/ptosis/epicanthus inversus syndrome, Dandy-Walker malformation, and global developmental delay. J Child Neurol 26(5):615–618
Darbro BW, Mahajan VB, Gakhar L, Skeie JM, Campbell E, Wu S, Bing X, Millen KJ, Dobyns WB, Kessler JA, Jalali A, Cremer J, Segre A, Manak JR, Aldinger KA, Suzuki S, Natsume N, Ono M, Hai HD, Viet le T, Loddo S, Valente EM, Bernardini L, Ghonge N, Ferguson PJ, Bassuk AG (2013) Mutations in extracellular matrix genes NID1 and LAMC1 cause autosomal dominant Dandy-Walker malformation and occipital cephaloceles. Hum Mutat 34(8):1075–1079
Basson MA, Wingate RJ (2013) Congenital hypoplasia of the cerebellum: developmental causes and behavioral consequences. Front Neuroanat 7:29
Sato T, Joyner AL (2009) The duration of Fgf8 isthmic organizer expression is key to patterning different tectal-isthmo-cerebellum structures. Development 136(21):3617–3626
Murray JC, Johnson JA, Bird TD (1985) Dandy-Walker malformation: etiologic heterogeneity and empiric recurrence risks. Clin Genet 28(4):272–283
De P, Carlson JH, Leyland-Jones B, Dey N (2015) Role of “oncogenic nexus” of CIP2A in breast oncogenesis: how does it work? Am J Cancer Res 5(9):2872–2891
Mirzaa GM, Campbell CD, Solovieff N, Goold C, Jansen LA, Menon S, Timms AE, Conti V, Biag JD, Adams C, Boyle EA, Collins S, Ishak G, Poliachik S, Girisha KM, Yeung KS, Chung BHY, Rahikkala E, Gunter SA, McDaniel SS, Macmurdo CF, Bernstein JA, Martin B, Leary R, Mahan S, Liu S, Weaver M, Doerschner M, Jhangiani S, Muzny DM, Boerwinkle E, Gibbs RA, Lupski JR, Shendure J, Saneto RP, Novotny EJ, Wilson CJ, Sellers WR, Morrissey M, Hevner RF, Ojemann JG, Guerrini R, Murphy LO, Winckler W, Dobyns WB (2016) Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol 73(7):836–845
Li H, Durbin R (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25(14):1754–1760
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22(3):568–576
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164
Gai N, Jiang C, Zou YY, Zheng Y, Liang DS, Novel WLQ (2016) SIL1 nonstop mutation in a Chinese consanguineous family with Marinesco-Sjogren syndrome and Dandy-Walker syndrome. Clin Chim Acta 458:1–4
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O'Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG, American College of Medical G, and Genomics (2013) ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 15(7):565–574
Dahlstrom KM, Salminen TA (2015) 3D model for cancerous inhibitor of protein phosphatase 2A armadillo domain unveils highly conserved protein-protein interaction characteristics. J Theor Biol 386:78–88
Kerosuo L, Bronner ME (2016) cMyc regulates the size of the premigratory neural crest stem cell pool. Cell Rep 17(10):2648–2659
Ventela S, Come C, Makela JA, Hobbs RM, Mannermaa L, Kallajoki M, Chan EK, Pandolfi PP, Toppari J, Westermarck J (2012) CIP2A promotes proliferation of spermatogonial progenitor cells and spermatogenesis in mice. PLoS One 7(3):e33209
Come C, Cvrljevic A, Khan MM, Treise I, Adler T, Aguilar-Pimentel JA, Au-Yeung B, Sittig E, Laajala TD, Chen Y, Oeder S, Calzada-Wack J, Horsch M, Aittokallio T, Busch DH, Ollert MW, Neff F, Beckers J, Gailus-Durner V, Fuchs H, Hrabe de Angelis M, Chen Z, Lahesmaa R, Westermarck J (2016) CIP2A promotes T-cell activation and immune response to listeria monocytogenes infection. PLoS One 11(4):e0152996
Waickman AT, Powell JD (2012) mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev 249(1):43–58
Acknowledgements
We are grateful to the patients and the family members.
Funding
CAY received grants from the Ministry of Science and Technology (MOST), Taiwan, grant 106-2314-B-039-047-MY3, and from China Medical University Hospital, Taiwan, grant DMR-107-205; JGC received grants from the China Medical University Hospital, Taiwan, grant DMR-106-230. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by the Ethics Review Board of China Medical University and Hospital in Taiwan (CMUH106-REC1-047).
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Yang, CA., Chou, IC., Cho, DY. et al. Whole exome sequencing in Dandy-Walker variant with intellectual disability reveals an activating CIP2A mutation as novel genetic cause. Neurogenetics 19, 157–163 (2018). https://doi.org/10.1007/s10048-018-0548-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-018-0548-6