
Abstract
Recessive mutations in genes of the glycosylphosphatidylinositol (GPI)-anchor synthesis pathway have been demonstrated as causative of GPI deficiency disorders associated with intellectual disability, seizures, and diverse congenital anomalies. We performed whole exome sequencing in a patient with progressive encephalopathies and multiple dysmorphism with hypophosphatasia and identified novel compound heterozygous mutations, c.250G>T (p. Glu84*) and c.1342C>T (p. Arg488Trp), in PIGT encoding a subunit of the GPI transamidase complex. The surface expression of GPI-anchored proteins (GPI-APs) on patient granulocytes was lower than that of healthy controls. Transfection of the Arg488Trp mutant PIGT construct, but not the Glu84* mutant, into PIGT-deficient cells partially restored the expression of GPI-APs DAF and CD59. These results indicate that PIGT mutations caused neurological impairment and multiple congenital anomalies in this patient.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glycosylphosphatidylinositol (GPI) acts as the anchor of various eukaryotic proteins expressed on the plasma membrane. GPI synthesis and GPI-anchored protein (GPI-AP) modification are mediated by at least 27 genes in the endoplasmic reticulum (ER) and Golgi apparatus [1]. Recent studies have indicated that inherited loss-of-function mutations in these genes lead to GPI deficiencies associated with neurological impairments including seizures, intellectual disability, and multiple congenital anomalies [2–9]. In addition, somatic mutations in PIGA cause paroxysmal nocturnal haemoglobinuria, a haematopoietic disease, which is also caused by somatic mutation of PIGT in combination with the germ line mutation of one allele [10, 11].
PIGT is one of the subunits of the GPI transamidase complex, and catalyzes the attachment of GPI anchors to proteins in the ER [1]. Kvarnung et al. [12] previously reported a homozygous PIGT mutation in patients from a consanguineous Turkish family with multiple congenital anomalies-hypotonia-seizures syndrome-3 (MCAHS3 [MIM 615398]). In the present study, we describe the use of whole exome sequencing to identify novel compound heterozygous PIGT mutations in a Japanese patient with seizures, intellectual disability and multiple congenital anomalies. Functional analysis indicated that these mutations are causative of GPI deficiency.
Patient and methods
Patient
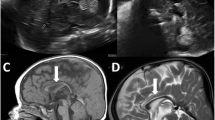
The female proband was born at full term without asphyxia as the first child of healthy unrelated parents (Fig. 1a). Polyhydramnios was recognized during pregnancy. She showed poor sucking and post-feed stridor soon after birth. At 4 months of age, she showed tonic seizures with apnea and myoclonic seizures, both of which repeatedly turned to convulsive status. Her electroencephalogram (EEG) demonstrated high-amplitude slow wave as a background activity, but no epileptic discharges were observed. She also showed a poor response, muscle hypotonia, unstable head control, a cardiac murmur caused by patent ductus arteriosus, and left hydronephroureter with ureteral stenosis. Her seizures were refractory to multiple antiepileptic drugs such as carbamazepine, clobazam, and an intravenous injection of pyridoxal phosphate while the frequency of her seizures decreased with the combination of valproic acid, zonisamide, and phenytoin to some extent. Phenobarbital could not be used in infancy because of drug eruption. After 1 year of age, she was frequently admitted to hospital because of convulsive status epilepticus induced by fever, or recurrent episodes of respiratory infections, bronchial asthma, or gastroenteritis. Her sleep cycle was disorganized. Brain magnetic resonance imaging at 3 years of age demonstrated progressive atrophy of the cerebral hemisphere, cerebellum, and brainstem (Fig. 1c). EEG at 3 years showed borderline findings consisting of a predominance of fast wave activity with no spindle formation interrupted by slow wave burst. She recurrently suffered bone fractures without obvious event. Systemic bone X-ray at 12 years of age showed neurogenic arthrogryposis and osteoporosis. At 12 years of age, she was bedridden and was only able to roll over. She showed profound intellectual disability and had no meaningful words. Her epileptic seizures disappeared after 10 years of age, but epileptic discharges comprised of spike-and-slow wave complex at bilateral frontal area with low-amplitude irregular background activity were seen on EEG.

a Familial pedigree. b Sanger sequencing results. Compound heterozygous mutations, c.250G>T and c.1342C>T, in PIGT were observed in the affected individual. c.250G>T (left) and c.1342C>T (right) were inherited from the mother and the father, respectively. c Magnetic resonance imaging of the patient’s brain. Axial and sagittal T1-weighted images at 3 years of age show atrophic changes of the cerebral hemisphere, brainstem, and cerebellum
G-banded chromosomal analysis revealed a normal karyotype (46,XX). Metabolic screenings including amino acids, lactic acid, pyruvic acid, organic acids, lactic acid, and lysosomal enzymes were unremarkable. The biochemical analysis of blood repeatedly showed low levels of serum alkaline phosphatase from birth (186 U/l at birth and 326 U/l at 7 years of age [normal range, 450–1250 U/l]). Both concentrations of serum and urine calcium were normal (serum calcium, 9.6 mg/dl; U-calcium/U-creatinine ratio, 0.23 at 7 years of age).
DNA preparation
Peripheral blood samples were obtained from the patient and her parents after parents signed informed consent. DNA was extracted using QuickGene-610 L (Fujifilm, Tokyo, Japan) according to the manufacturer’s instructions. The study was approved by the ethics committee of the Yokohama City University.
Whole exome sequencing
Patient DNA was captured with the SureSelect Human All Exon V5 Kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina Hiseq2000 (Illumina, San Diego, CA, USA) with 101-bp paired-end reads. Image analysis and base calling were performed by sequence control software real-time analysis and CASAVA software v1.8 (Illumina). Reads were mapped to the human reference genome sequence (UCSC hg19, NCBI build 37) and aligned using Novoalign (Novocraft Technologies, Jaya, Malaysia). PCR duplicate reads were excluded using Picard (http://picard.sourceforge.net/) for further analysis. Single-nucleotide variants (SNVs) and small indels were identified using the Genome Analysis Toolkit UnifiedGenotyper [13] and filtered according to the Broad Institute’s best-practice guidelines (version 3). Variants that passed the filters were annotated using ANNOVAR[14]. The damaging prediction was performed by Polyphen-2 [15] and MutationTaster software [16].
Sanger sequencing
PIGT exon 2 and exon 10 sequences were PCR amplified from the patient and her parents using the following primers: PIGT ex2F 5′-GGGAGGAACTTGTCATCACC-3′ and ex2R 5′-CAGTGGCAGGATGACAACAC-3′, PIGT ex10F 5′-AGAGATGTGGGTGACCTTGC-3′ and ex10R 5′-CTGAGGACAGATGGGCTACA-3′, respectively. Amplified PCR products were sequenced on an ABI 3500xl or 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Flow cytometry
Peripheral blood samples were collected from the patient and normal control individuals. Granulocyte surface expression of total GPI-APs was quantified by staining with Alexa 488-conjugated inactivated aerolysin (FLAER; Protox Biotech, Victoria, Canada). Expression of CD16, CD24, and alkaline phosphatase (ALP) was examined using appropriate primary antibodies (3G8, ML5, and B4-78, respectively; BD Biosciences, Franklin Lakes, NJ, USA), followed by a PE-conjugated anti-mouse IgG secondary antibody (BD Biosciences). Cells were analyzed by BD FACSCanto II (BD Biosciences).
Human PIGT cDNA (NM_015937.5) with FLAG at the C terminus was subcloned into the pME (driven by a strong SRα promoter) or pTA (driven by a weak promoter containing only TATA-box) vector [17]. Two PIGT mutants, Glu84* and Arg488Trp, were generated by site-directed mutagenesis. Mutant and wild-type PIGT plasmids were transfected by electroporation into CHO H4, PIGT-deficient Chinese hamster ovary (CHO) cells expressing human DAF (also called CD55) and CD59 as previously described [18]. Two days later, lysates were run on SDS-PAGE, and Western blotting was performed using an anti-FLAG antibody (M2; Sigma-Aldrich, St. Louis, MO, USA) to detect FLAG-tagged PIGT (PIGT-F). The protein levels were normalized to the loading control, and luciferase activities were used to evaluate transfection efficiencies. Cells were stained with anti-hCD59 (5H8), anti-hDAF (IA10), and anti-Hamster uPAR (5D6) antibodies and restoration of the surface expression of GPI-APs was assessed by flow cytometry.
Results
Mutation screening
We performed mutation screening for previously reported genes involved in the GPI-anchor–synthesis pathway, and identified the compound heterozygous mutations c.250G>T (p. Glu84*) and c.1342C>T (p. Arg488Trp) in PIGT (NM_015937.5). Both mutations were not found in 6500 ESP (Exome Sequencing Project) or 1000 genomes [19, 20], but c.1342C>T is present in one of 408 in-house control exomes. Both mutations were predicted to be probably disease-causing by Polyphen-2 and MutationTaster. Sanger sequencing confirmed that c.250G>T and c.1342C>T were inherited from the mother and father, respectively (Fig. 1b).
Functional effect of the mutations on GPI synthesis
PIGT is a component of GPI transamidase that mediates the post-translational attachment of GPI anchors to the C-terminal of the precursor protein. Therefore, the mutant GPI transamidase is likely to impair the surface expression of GPI-APs. To investigate the influence of PIGT mutations on GPI-APs synthesis, we first examined the granulocyte surface expression of GPI-APs from the patient and a healthy control. Expression of total GPI-APs (FLAER staining) and GPI-APs CD16 and ALP on granulocytes was reduced in the patient compared to the normal control (Fig. 2a). However, similar expression levels of another GPI-AP CD24 were seen in the patient and control (Fig. 2a).

a Surface expression of GPI-APs on granulocytes. Granulocytes from the patient and healthy control were stained with FLAER or antibodies against CD24, CD16, and ALP. The expression of total GPI, CD16, and ALP in the patient (solid line) was lower than in the normal control (dark shaded area). CD24 expression did not differ between the patient and control. The light shaded areas represent the isotype control. X axes show fluorescent intensities, which indicate expression levels of each GPI-AP on the cell surface. Y axes show the relative cell numbers. The value of mean fluorescent intensities of each sample is shown in each panel. b PIGT-deficient CHO cells were transiently transfected with wild-type (dashed line), Glu84* mutant (fine solid line), or Arg488Trp mutant (bold solid line) PIGT cDNA expression constructs in vectors with either a strong promoter (pME; upper panels) or weak promoter (pTA; lower panels). PIGT-F protein levels and restoration of the surface expression of CD59, DAF, and uPAR were assessed 2 days later. The dark and light shadows represent empty-vector transfectants and isotype controls, respectively. c Western blotting showed that the Arg488Trp mutant protein was expressed at similar levels to the wild-type protein, whereas the Glu84* mutant full-length protein, representing the read-through product, was expressed at lower levels. Quantity numbers at the bottom of the gel indicate the relative intensity of PIGT-F protein levels normalized to the loading control, and luciferase activities used for evaluating transfection efficiencies. Arrowhead indicates a non-specific product
We then transiently transfected wild-type or mutant (Glu84* or Arg488Trp) PIGT cDNA constructs into PIGT-deficient CHO cells to evaluate the functional effect of each mutation on GPI-AP expression. Western blotting revealed that the expression level of Arg488Trp mutant protein was similar to that of wild-type protein, whereas the Glu84* mutant expressed a small amount of full-length protein (probably read-though) (Fig. 2c). Wild-type PIGT transfection successfully restored the expression of GPI-APs CD59, DAF (CD55), and uPAR in both cases using vectors with a strong (pME) and weak (pTA) promoter (Fig. 2b). The Arg488Trp mutant PIGT cloned in pME restored the expression of GPI-APs close to that of wild-type, whereas the same mutant in the pTA vector only partially restored expression. The Glu84* mutant PIGT in the pME vector insufficiently restored the expression of GPI-APs, while this mutant in the pTA vector could not restore expression (Fig. 2b). These results demonstrate that both mutants, especially the Glu84* alteration, reduce the activity of PIGT function.
Discussion
GPI deficiency syndromes are recessive disorders caused by mutations in genes involved in the GPI-anchor biosynthesis pathway. Here, we describe novel compound heterozygous PIGT mutations in a nonconsanguineous patient presenting with seizures and intellectual disability.
The first reported PIGT mutation (c.547A>C, p.Thr183Pro) was identified in a consanguineous Turkish family who showed seizures, intellectual disability, and multiple congenital anomalies [12]. A decreased expression of GPI-APs was documented on patient granulocytes. They confirmed that the homozygous c.547A>C mutation impaired the function of PIGT by the functional study using pigt knockdown zebrafish embryos which showed gastrulation defects phenotype. In the present study, we also demonstrated that both PIGT mutations, c.250G>T (p. Glu84*) and c.1342C>T (p. Arg488Trp), impaired the function of PIGT which was confirmed by the functional study using the PIGT deficient CHO cells.
Mammalian GPI transamidase consists of at least five subunits, PIGK, GPAA1, PIGS, PIGT, and PIGU [1]. Of these, PIGT plays a critical role in stabilizing the complex formation of GPI transamidase [17], which mediates cleavage of the GPI attachment signal peptide at the C-terminal of the precursor protein and transfers GPI anchors to the C-terminal of cleaved proteins [1]. Consequently, PIGT mutants may not be able to correctly form the GPI transamidase complex, leading to a loss of GPI transamidase activity and reduction in the cellular surface expression of GPI-APs.
Our patient and four patients described by Kvarnung et al. [12] showed broad clinical spectrum and shared several common features (Table 1). The neurological findings including intractable seizures, hypotonia and severe intellectual disability were observed in all patients. Ophthalmologic features including strabismus, nystagmus, and cerebral visual impairment were also observed in all. Cerebral and cerebellar atrophy was observed in our patient and two of four seen by Kvarnung et al. The EEG findings in our patient were also exacerbated as she grew, suggesting progressive encephalopathy. Our patient and three of four patients by Kvarnung et al. had some cardiologic disorders. All patients had some urologic features, but not nephrocalcinosis in our patient. Our case shared similar facial features with previous patients including a depressed nasal bridge, short anteverted nose, tented lip, and downturned corners of the mouth. Low set ears, micrognathia, malar flattening, and upslanting palpebral fissures were unique to our patient.
Hyperphosphatasia is a characteristic symptom of some GPI deficiencies, such as PIGV, PIGW, PIGO, PGAP2 and PGAP3 deficiencies [2–6]. In contrast, hypophosphatasia is a particularly distinctive feature in the loss of GPI transamidase function. Murakami at al. suggested that GPI transamidase abnormalities lead to an inability to hydrolyze the precursor protein of alkaline phosphatase, resulting in the degradation of most precursor proteins within the cell and a decrease of serum alkaline phosphatase levels (hypophosphatasia) [21]. This is supported in our case by the hypophosphatasia. The patients described by Kvarnung et al. showed hypercalcemia and hypercalciuria following tooth abnormality, craniosynostosis, a delayed bone age, and reduced mineralization, which is the common features with infantile hypophosphatasia caused by the mutations in ALPL, the gene encoding tissue non-specific alkaline phosphatase (TNAP) [22]. As TNAP is a GPI-AP, the PIGT deficiency causes decreased surface expression of TNAP, which would lead to bone abnormalities. Regardless of hypophosphatasia, our case showed only mild scoliosis and osteoporosis, but no tooth abnormality nor craniosynostosis. Different mutational effects on the enzyme activity may account for such different phenotypes. In this study, mutant PIGT construct harboring Arg488Trp or Glu84* in strong promoter (pME) vector restored GPI-Aps expression. In contrast, Kvarnung et al. showed that abnormal phenotype of pigt knockdown zebrafish was never restored by the homozygous mutant (Thr183Pro) PIGT cDNA. Therefore, it is possible to estimate that the Thr183Pro mutation may affect the GPI transamidase complex activity more severely than the Arg488Trp and Glu84* mutations, leading to less severe phenotypes. However, further functional analysis and cases with PIGT mutations are needed to elucidate the relevance of these mutations in PIGT function and full clinical spectrum of GPI deficiency syndromes.
References
Kinoshita T, Fujita M, Maeda Y (2008) Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: recent progress. J Biochem 144(3):287–294. doi:10.1093/jb/mvn090
Krawitz PM, Schweiger MR, Rodelsperger C, Marcelis C, Kolsch U, Meisel C, Stephani F, Kinoshita T, Murakami Y, Bauer S, Isau M, Fischer A, Dahl A, Kerick M, Hecht J, Kohler S, Jager M, Grunhagen J, de Condor BJ, Doelken S, Brunner HG, Meinecke P, Passarge E, Thompson MD, Cole DE, Horn D, Roscioli T, Mundlos S, Robinson PN (2010) Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet 42(10):827–829. doi:10.1038/ng.653
Krawitz PM, Murakami Y, Hecht J, Kruger U, Holder SE, Mortier GR, Delle Chiaie B, De Baere E, Thompson MD, Roscioli T, Kielbasa S, Kinoshita T, Mundlos S, Robinson PN, Horn D (2012) Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet 91(1):146–151. doi:10.1016/j.ajhg.2012.05.004
Hansen L, Tawamie H, Murakami Y, Mang Y, ur Rehman S, Buchert R, Schaffer S, Muhammad S, Bak M, Nothen MM, Bennett EP, Maeda Y, Aigner M, Reis A, Kinoshita T, Tommerup N, Baig SM, Abou Jamra R (2013) Hypomorphic mutations in PGAP2, encoding a GPI-anchor-remodeling protein, cause autosomal-recessive intellectual disability. Am J Hum Genet 92(4):575–583. doi:10.1016/j.ajhg.2013.03.008
Howard MF, Murakami Y, Pagnamenta AT, Daumer-Haas C, Fischer B, Hecht J, Keays DA, Knight SJ, Kolsch U, Kruger U, Leiz S, Maeda Y, Mitchell D, Mundlos S, Phillips JA 3rd, Robinson PN, Kini U, Taylor JC, Horn D, Kinoshita T, Krawitz PM (2014) Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. Am J Hum Genet 94(2):278–287. doi:10.1016/j.ajhg.2013.12.012
Chiyonobu T, Inoue N, Morimoto M, Kinoshita T, Murakami Y (2013) Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with West syndrome and hyperphosphatasia with mental retardation syndrome. J Med Genet. doi:10.1136/jmedgenet-2013-102156
Krawitz PM, Murakami Y, Riess A, Hietala M, Kruger U, Zhu N, Kinoshita T, Mundlos S, Hecht J, Robinson PN, Horn D (2013) PGAP2 mutations, affecting the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation syndrome. Am J Hum Genet 92(4):584–589. doi:10.1016/j.ajhg.2013.03.011
Almeida AM, Murakami Y, Layton DM, Hillmen P, Sellick GS, Maeda Y, Richards S, Patterson S, Kotsianidis I, Mollica L, Crawford DH, Baker A, Ferguson M, Roberts I, Houlston R, Kinoshita T, Karadimitris A (2006) Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 12(7):846–851. doi:10.1038/nm1410
Ng BG, Hackmann K, Jones MA, Eroshkin AM, He P, Wiliams R, Bhide S, Cantagrel V, Gleeson JG, Paller AS, Schnur RE, Tinschert S, Zunich J, Hegde MR, Freeze HH (2012) Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet 90(4):685–688. doi:10.1016/j.ajhg.2012.02.010
Johnston JJ, Gropman AL, Sapp JC, Teer JK, Martin JM, Liu CF, Yuan X, Ye Z, Cheng L, Brodsky RA, Biesecker LG (2012) The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 90(2):295–300. doi:10.1016/j.ajhg.2011.11.031
Krawitz PM, Hochsmann B, Murakami Y, Teubner B, Kruger U, Klopocki E, Neitzel H, Hoellein A, Schneider C, Parkhomchuk D, Hecht J, Robinson PN, Mundlos S, Kinoshita T, Schrezenmeier H (2013) A case of paroxysmal nocturnal hemoglobinuria caused by a germline mutation and a somatic mutation in PIGT. Blood 122(7):1312–1315. doi:10.1182/blood-2013-01-481499
Kvarnung M, Nilsson D, Lindstrand A, Korenke GC, Chiang SC, Blennow E, Bergmann M, Stodberg T, Makitie O, Anderlid BM, Bryceson YT, Nordenskjold M, Nordgren A (2013) A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 50(8):521–528. doi:10.1136/jmedgenet-2013-101654
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. doi:10.1038/ng.806
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164. doi:10.1093/nar/gkq603
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. doi:10.1038/nmeth0410-248
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7(8):575–576. doi:10.1038/nmeth0810-575
Ohishi K, Inoue N, Kinoshita T (2001) PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J 20(15):4088–4098. doi:10.1093/emboj/20.15.4088
Ashida H, Hong Y, Murakami Y, Shishioh N, Sugimoto N, Kim YU, Maeda Y, Kinoshita T (2005) Mammalian PIG-X and yeast Pbn1p are the essential components of glycosylphosphatidylinositol-mannosyltransferase I. Mol Biol Cell 16(3):1439–1448. doi:10.1091/mbc.E04-09-0802
Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA (2010) A map of human genome variation from population-scale sequencing. Nature 467(7319):1061–1073. doi:10.1038/nature09534
Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491(7422):56–65. doi:10.1038/nature11632
Murakami Y, Kanzawa N, Saito K, Krawitz PM, Mundlos S, Robinson PN, Karadimitris A, Maeda Y, Kinoshita T (2012) Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J Biol Chem 287(9):6318–6325. doi:10.1074/jbc.M111.331090
Mornet E (2007) Hypophosphatasia. Orphanet J Rare Dis 2:40. doi:10.1186/1750-1172-2-40
Acknowledgments
We thank the patient’s family for participating in this work. We also thank Nobuko Watanabe for her technical assistance. This study was supported by the Ministry of Health, Labour and Welfare of Japan, a Grant-in-Aid for Scientific Research (A), (B), and (C) from the Japan Society for the Promotion of Science, the Takeda Science Foundation, the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems, the Strategic Research Program for Brain Sciences, and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nakashima, M., Kashii, H., Murakami, Y. et al. Novel compound heterozygous PIGT mutations caused multiple congenital anomalies-hypotonia-seizures syndrome 3. Neurogenetics 15, 193–200 (2014). https://doi.org/10.1007/s10048-014-0408-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-014-0408-y