
Abstract
It has been found that CDKL5 gene mutations are responsible for early-onset epilepsy and drug resistance. We screened a population of 92 patients with classic/atypical Rett syndrome, 17 Angelman/Angelman-like patients and six idiopathic autistic patients for CDKL5 mutations and exon deletions and identified seven novel mutations: six in the Rett subset and one in an Angelman patient. This last, an insertion in exon 11, c.903_904 dupGA, p.Leu302Aspfx49X, is associated with a relatively mild clinical presentation as the patient is the only one capable of sitting and walking alone. Of the six mutations, two are de novo missense changes affecting highly conserved aminoacid residues, c.215 T > C p.Ile72Thr and c.380A > G p.His127Arg (present in a mosaic condition) found in two girls with the most severe clinical presentation, while the remaining are the splicing c.145 + 2 T > C and c.2376 + 5G > A, the c.1648C > T p.Arg550X and the MPLA-identified c.162_99del261 mutation. RNA characterisation of four mutations revealed the aberrant transcript of the missense allele (case 2) and not the stop mutation (case 3), but also allowed the splicing mutation (case 1) and the c.-162_99del261 (case 4) to be categorised as truncating. The obtained data reinforce the view that a more severe phenotype is due more to an altered protein than haploinsufficiency. Furthermore, the mutational repertoire of the CDKL5 gene is shown to be expanded by testing patients with phenotypical overlap to Rett syndrome and applying multiplex ligation-dependent probe amplification.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Following the description of two girls with West syndrome (OMIM#308350) carrying X;autosome translocations disrupting the cyclin-dependent kinase-like 5 gene (CDKL5; OMIM#300203) [1], other mutations within the same gene have been reported in patients manifesting a similar clinical picture, thus indicating that the associated phenotype is characterised by early-onset untreatable seizures, infantile spasms, severe mental retardation and some features of Rett syndrome (RTT: OMIM#312750) [2–6]. On the basis of this clinical presentation, the CDKL5-positive group has been assimilated to the Hanefeld early seizure variant of RTT [2]. Bahi-Buisson [7] has recently identified an epilepsy phenotype whose three-stage pattern begins with “early epilepsy”, moves on to epileptic encephalopathy and ends with late multifocal and myoclonic epilepsy.
The human CDKL5 gene product, also called serine/threonine kinase 9 (STK9), belongs to the serine–threonine kinase family, which shares homology with members of the mitogen-activated protein and cyclin-dependent kinase (CDK) families [8]. CDKL5 is a ubiquitous protein that is mainly expressed in the brain, testes and thymus [9], shuttling between the nucleus and cytoplasm and varying in different brain areas and during development [10]. Its subcellular distribution seems to be modulated by its C-terminal tail, which is responsible for an active nuclear export mechanism [10].
The clinical overlap between CDKL5-mutated patients and patients with RTT, which is caused by mutations in the Methyl-CpG-binding protein 2 gene (MeCP2: OMIM#300005), prompted expression and functional studies demonstrating that CDKL5 and MeCP2 belong to the same pathway [11, 12]. The kinase activity of CDKL5, which bears the catalytic site in the aminoterminal region, has been confirmed by the in vitro kinase assay and the fact that it phosporylates MECP2, probably through intermediate signalling interactors [9, 11]. These studies have also documented the spatial and temporal overlapping of Mecp2 and Cdkl5 expression profiles in embryonic and post-natal mouse brain [9, 11]. Functional characterisations of human CDKL5 mutations in both the catalytic and the large C terminus indicate that the COOH-terminal domain negatively influences kinase self-phosphorylation and cooperates with the catalytic domain in regulating the nuclear localisation of the kinase [9, 12].
Thirty-eight CDKL5 mutations have so far been described [2, 6, 7, 13, 14], and recently, various missense mutations have been found in three unrelated boys affected by profound mental retardation and intractable seizures, thus suggesting that amino acid changes in the CDKL5 protein can be tolerated despite the hemizygous condition [15]. Given the small number of patients, genotype–phenotype correlation studies have been limited, but it has been suggested that mutations affecting the aminoterminal catalytic domain are associated with a more severe phenotype consisting of an earlier onset and intractable infantile spasms followed by late-onset multifocal and myoclonic epilepsy [7]. A more extensive knowledge of the currently restricted CDKL5 mutational repertoire might provide new clues concerning genotype–phenotype correlations, and analysing synthetic derivatives of particular mutants in functional assays might highlight new protein domains.
We here describe seven novel CDKL5 mutations detected by screening patients with MeCP2-negativeRett syndrome, Angelman/Angelman-like syndrome (AS), or idiopathic autism (ADS). The sequence alterations include whole-exons CDKL5 deletion first revealed by means of multiplex ligation-dependent probe amplification (MLPA) and two mutations mapping to rarely affected kinase regions near to and downstream of the NH2 domain, the former carried by an Angelman patient. These findings evidence that the mutational repertoire of CDKL5 and the associated phenotypes remain to be further disclosed.
Materials and methods
Patients
One hundred and fifteen patients with precocious epilepsy (17 males), including 92 with classic or atypical Rett syndrome, 17 with Angelman or Angelman-like syndrome and six autistic patients, were recruited from local Childhood Neuropsychiatry Divisions. The Rett syndrome cohort mainly included patients with the classic or the congenital form, negative to MECP2 test. The Angelman subset consisted of patients presenting with most major and a few minor criteria for the syndrome (16) who proved negative to methylation assay and UBE3A scan. Autism was diagnosed using the ADI/ADOS interview. All of the patients were characterised by early-onset seizures, and most patients (including the CDKL5 mutation carriers described below) were carefully examined by means of serial EEG and evoked potential recordings and brain MRI.
Molecular investigations
Blood samples were collected after informed consent was signed by the patients′ parents. MeCP2e2 mutations were screened by means of direct sequencing using an automated sequencer (ABI PRISM 3100, PE Applied Biosystem, Foster City, CA, USA) and intronic exon-flanking primers with fluorescent dye terminators designed by Primer Express software (available on request); exon 1 of the MeCP2e1 isoform was sequenced as suggested by Mnatzakanian et al. [17].
The Angelman/Angelman-like patients underwent methylation testing by means of M-PCR [18] and UBE3A mutational screening and direct sequencing of the coding region. The primers were chosen using Primer Express software (Applied Biosystem).
For CDKL5 mutational screening, the coding and the neighbouring intronic regions were analysed using previously described primers and conditions [19]. The PCR products were loaded onto a Transgenomic WAVE denaturing high-performance liquid chromatograph, and those showing abnormal chromatograms were sequenced on an ABI PRISM 310.
The MECP2 and CDKL5 MLPA kit (MRC-Holland, Amsterdam, NL) was used in accordance with the manufacturer′s instructions.
The methylation status of the CDKL5-mutated patients was assessed using the Humara Androgen Receptor gene methylation assay as previously described [20]. The triplet repeats were separated by means of capillary electrophoresis and analysed using an ABI PRISM 310. Mutations within exon 2 of the Aristaless gene (ARX) were investigated using Evans′ protocol [21].
For control samples analysis was performed on peripheral blood DNA.
cDNA analysis of the mutations
Total RNA was extracted from fresh peripheral blood or lymphoblastoid cell lines using the Trizol procedure (Life Technologies, Grand Island, NY, USA), and cDNA was obtained using the SuperScriptTM First-Strand Synthesis System (Invitrogen Ltd, Inchinnan Business Park, UK) for RT-PCR, with the primers being selected by Primer Express software. Primers amplifying a product from exon 2 to exon 6 (fw: GCCATGTTTTGTGGCTTGCAT and rev: GCTTTTTACTTTCTCAGGT) were used to characterise the splicing mutation; primers (fw AGAAACGACTTTACGAGAG (ex 5) and rev GCTTTTTACTTTCTCAGGT (ex 6)) were designed for the missense mutation; and the fw ACTCCAAGTCTGTGAGCAAC and rev CTATGCTCAAGCTTCCATC primers in exon 12 were used for the truncating mutation. Primers GTCCCCTGCTAGAAGTGG within the 5′ UTR exon 1b and rev tcattttcttcactgtccttg in exon 3 were used for the MPLA deletion aminoterminal of isoform NM_003159.
The PCR products obtained from the cDNAs were loaded on 3% agarose gel, eluted by GFX (Gel Heathcare, Buckinghamshire UK) and sequenced using the above primers in order to evaluate the presence of the aberrant transcripts. A real-time qPCR assay was performed with the comparative threshold cycle method and Syber Green protocol (SYBR GREEN, PCR Master Mix, Applied Biosystem, Warrington, UK) by using primer pairs designed by the Primer Express software and replicated using TaqMan probes: Hs01043900m1 (CDKL5 C-terminal sequence) and Hs99999909 (HPRT1 ex6-7).
CDKL5 expression levels were determined on three independent RNA preparations and triplicated readings each, for both the test and control samples and were compared to the internal HGPRT control gene. The threshold cycle number (Ct) was determined for all of the PCRs, and a comparative Ct method [22, 23] was used to calculate the related number of alleles. The relative gene copy number was calculated by means of the expression 2(ddCt+/-2 s), which represents the standard deviation (SD) of the difference calculated from the Ct SD of both CDKL5 and HPRT.
Results
Ninety-two patients (including six males) characterised by the onset of seizures in their first year of life and refractoriness to medications were selected from a sample of 180 patients with Rett/Rett-like phenotype, found to be negative at MeCP2 mutation analysis (point mutations, gross and intragenic deletions) and enrolled for CDKL5 mutational screening, together with 17 Angelman/Angelman-like patients (six males) who were negative for all of the known causes of 15q11.2 molecular anomalies and presented with very early seizures and six epileptic children (five males) diagnosed as autistic.
CDKL5 scan revealed five novel sequence alterations and one two-exon deletion within the coding region in the Rett patients and one novel insertion in an Angelman patient. None of the six autistic children showed any alteration in the CDKL5 coding sequence. The sequencing of exon 2 of the Aristaless homeobox ARX did not reveal any mutation in any of the CDKL5-negative cases.
Clinical evaluation of CDKL5 mutation carriers
Table 1 summarises the clinical and neurological data relating to the seven mutated patients and includes most of the age-related clinical signs so far reported in CDKL5 patients. All seven patients are females, had experienced a normal perinatal period, had a normal head circumference at birth and developed deceleration and microcephaly within the first 2 years (except for no. 6, who did not develop microcephaly and no. 7 who is <1 year old). They all manifest severe intellectual disability, very limited hand skills, hypotonia, lack of eye contact and the absence of speech and walking (except for 5), but only patients 6 and 7 have undergone a regressive stage.
Their seizures, which appeared by the second week of life, were characterised by generalised activity with multifocal spikes and without hypsarrhythmia at onset. Three patients, 2, 4 and 5, conformed to the reported trend of pharmacological resistance, whereas no. 1, who was initially drug resistant, proved to be sensitive to topiramate and has been seizure free for 2 years; no. 3 is fairly controlled by drugs; no. 6 has slightly improved since she started receiving levetiracetam; lastly, in no. 7, seizures disappeared spontaneously at 6 months, and she is currently fully controlled and out of medication. MRI was abnormal in four of six tested patients. Additional clinical data concerning patients 1 and 3 have been reported by Pintaudi [14]. A few clinical traits were individual. Patient 2 experienced precocious puberty when she was 5 years old, and she and no. 6 are the only two who are not yet able to sit unaided. Patient 4 did not present either autonomic features or hand stereotypies, although the deliberate use of her hands is quite poor. Patient 5 is the first reported with features of Angelman syndrome: She displays all the consistent Angelman diagnostic criteria [16], i.e. absence of speech, severe developmental delay, ataxic gait, hypermotoric behaviour and easily excitable personality with uplifted hand-flapping rather than washing. She is microcephalic and shows intractable generalised tonic clonic and partial complex seizures, characterised by the slowing of background intermittent spikes. Her facies is typical of Angelman syndrome for brachycephaly, wide mouth and widely dispersed teeth and progressive prognatism. She is also autistic, but has relatively better motor skills than the other CDKL5-mutated girls (she can walk) and has infrequent seizures. Patient 6 is very severe and also shows cortical blindness. Early partial complex seizures with focal EEG changing into multifocal with developmental arrest, normal MRI and normal extensive metabolic workup brought us to the diagnosis of no.7.
Molecular characterisation of CDKL5 mutations
Patient 1 carries a novel splice mutation, c.145 + 2 T > C (Fig. 1a) that was not found in either parent and affects the splice donor site of exon 4. In addition to the expected 324 bp band, amplification of cDNA from exons 2 to 6 revealed a lower MW band of 278 bp (Fig. 1c). The aberrant transcript seems to be well represented as shown by the intensity of the band on agarose gel and the peak control/mutant ratio when the PCR products are separated by capillary electrophoresis (not shown), which is consistent with random X inactivation (50%). The sequencing of this aberrant band showed exon 4 skipping (Fig. 1b), which indicates that the alteration in the IVS4 donor site probably makes the IVS3 donor site refer directly to the IVS4 acceptor without using the IVS3 acceptor (Fig. 1d). The new junction predicts an alteration in the translational frame as a result of the introduction of 59 aberrant amino acids and a stop codon after Lys33 leading to premature truncation of the protein, p.Glu34LysfxX60.

Electropherograms of the splicing c.145 + 2 T > C mutation found in patient 1; on peripheral blood genomic (a) and cDNA(b); (c) agarose run evidencing both mutant (M) and control (C) transcripts; (d) putative splicing mechanism allowing the exclusion of exon 4
Patient 2 carries a de novo T > C substitution, c.215 T > C p.Ile72Thr, affecting nt 215 of the coding sequence and leading to the replacement of isoleucine72 by threonine (Fig. 2a). A high degree of evolutionary conservation is shown for Ile72 by protein sequence alignment (data not shown). Analysis of the cDNA revealed a small peak for the mutant allele (Fig. 2b), although X inactivation seemed to be balanced and reproducible also in the lymphoblastoid cell line (data not shown). Quantification in the latter of the amount of CDKL5 by real-time PCR showed a transcript level only slightly lower as compared to the control (data not shown)

Genomic (a) and cDNA (b) electropherograms of the missense c.215 T > C p.Ile72Thr mutation
Patient 3 carries a de novo c.1648C > T p.Arg550X mutation that predicts the conversion of Arg 550 to a stop codon leading to a truncated protein. In accordance with the rule that an aberrant transcript carrying a stop codon more than 50 bp from the splicing site undergoes mRNA-mediated decay, cDNA analysis showed that the mutated allele was not represented as only the normal allele was detected (data not shown). Real-time PCR on the lymphoblastoid cell line, which showed a balanced X inactivation likewise the peripheral blood, demonstrated a 50% level of transcript when compared to control, so confirming the decay of the mutant mRNA.
In patient 4, the mutation c.-162_99del261 was identified by means of the MLPA test, which was applied to all CDKL5-negative patients in order to detect intragenic deletions/duplications. Normalisation of the peak areas identified halved values for the probes corresponding to exons 2 and 3 (Fig. 3a), and PCR of cDNA using a primer mapping within the 5′ UTR exon of the NM_003159 isoform and the other downstream exon 4 revealed a deletion of 261 bp (Fig. 3b). Sequencing of the aberrant transcript confirmed that the whole sequence of exons 2 and 3 was missing (Fig. 3c). This deletion does not alter the reading frame, but removes the original ATG. Several ATGs are present in the newly formed sequence, but all predict the creation of reading frames with premature stop codons.

a MLPA results: The two bars corresponding to the deleted exons 2 and 3 are arrowed; b electrophoresis of peripheral blood cDNA (lane 1 patient, M, lanes 2 and 3 controls, C) shows in the patient the deleted (119 bp) and the expected (380 bp) bands; c cDNA sequencing defines exactly the deletion, joining the last base of exon 1 to the first base of exon 4
In patient 5, a de novo insertion of two nt was identified in exon 11, c.903_904 dupGA, which causes a stop codon 49 amino acids downstream, p.Leu302Aspfx49X (data not shown). No RNA was available precluding cDNA analysis.
In patient 6, an A > G transition was detected in exon 6 at nt 380, leading to the replacement of His127 by Arg (p.His127Arg). The affected residue, which shows a high degree of evolutionary conservation (not shown), belongs to the active serine–threonine kinase site. The sequence alteration was not found in either parent and >100 X chromosomes from normal individuals and was not observed in any of the other patients enrolled in this study. On the basis of its peak height (Fig. 4), it seems to be in a mosaic condition. We found the same electropherogram profile on fibroblasts’ DNA (data not shown). No RNA was available for transcript analysis.

Mosaicism of c.380A > G mutation is evidenced by the different height of peaks visible both in forward (a) and reverse (b) sequence
In patient 7, a G > A transition was found in the fifth nucleotide of the acceptor consensus of IVS16 c.2376 + 5G > A (supplementary figure on line); splice view software reveals that the consensus sequence is abolished by the substitution, thus leading to missplicing. The de novo origin of the mutation was confirmed, and we could rule out the observed alteration in 100 controls. RNA was not available for further studies.
A number of SNPs were detected and are summarised in Supplementary Table 2 (online), including an unreported coding c. 2767C > T p.Arg923Cys, and the previously reported coding p.Gln791Pro variant [22]. A patient carrying two silent substitution, p.Hys1001His and p.Thr1028Thr, in exon 21 plus the intronic variant c.404-53 T > C in IVS6 was found. All the polymorphisms were maternally inherited; evaluation of X inactivation showed that all are balanced. Additional five intronic and one 3′ UTR variant are shown, but their origin could not be established because of parental refusal.
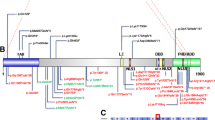
Figure 5 provides an updated overview of all of the published (including balanced) translocations found to disrupt the CDKL5 gene [23] and our CDKL5 mutations. Mutations span the whole gene and in terms of the types of mutation, 21 are truncating, ten are splice site and 13 are missense mutations, of whom three in male patients [15]. A mutation hotspot is apparent at codon 172, where we identified a novel change (p.Ile72Thr) in patient 2, while the p.His127Arg carried by our patient 6 in a mosaic condition is the only missense mutation in the serine–threonine kinase domain. Our case 4 mutation, involving the deletion of two whole exons and the ATG codon, is difficult to classify.

Schematic representation of the CDKL5 gene with its 21 exons. All the mutations reported in the literature are placed in the proper position on the gene, including those (in blue) recently identified in males. Truncating and splice mutations are indicated with cDNA nomenclature and missense mutations with protein nomenclature. Mutations detected by this study are in purple . At the bottom , schematic of the CDKL5 protein with its known functional domains (11)
Discussion
We screened CDKL5 mutations in patients with three neurodevelopmental disorders (RTT, AS and ADS) sharing the common features of early-onset and drug-resistant seizures.
AS and AS-like enter in differential diagnosis with RTT, a phenotypical overlap which was first indicated by the finding of five MeCP2 mutated cases (four females and one male) in a cohort of 47 AS patients (25 females and 22 males) with no molecular 15q11-13 anomaly [24]. A few studies have recommended extending CDKL5 screening to patients with a shared clinical phenotype, including AS and AS-like patients [19, 22], but no mutated cases have been previously identified. The patient described by Tao carrying a p.Cys152Phe was also tested for methylation pattern, but she has a clinical picture more severe than an Angelman patient [22]. Our patient 5, who fulfilled the main criteria for AS [16], is the first shown to carry a CDKL5 gene mutation. The finding of CDKL5 mutations in two cases with an Angelman-like phenotype supports the view that the screening of CDKL5 in such patients might be successful. Our decision to include a few autistic patients fulfilling the inclusion criterion of early-onset epilepsy was based on the shared autistic traits of AS and ADS cases and the reported finding of a CDKL5 mutation in one autistic patient [13]. A larger sample of autistic patients with early-onset seizures needs to be examined in order to establish the involvement of CDKL5 in this subset.
Our exhaustive molecular analysis not only included sequencing the whole coding sequence and exon-flanking regions but also the MLPA screening of whole exon deletions/duplications in a subgroup of patients with negative CDKL5 scan results, which identified one deleted patient (case 4) and indicates that at least a few CDKL5 mutations are not detected by standard mutational screening methods. The frequency of CDKL5 mutations in the selected cohorts is 7.6%, concordant with that reported by Bahi-Buisson (8.1%) for patients with refractory epilepsy [6], which has been the main selection criterion for our cohorts and much less than that found for children with infantil spasms (16.7%) [6].
Our findings increase the relatively restricted repertoire of CDKL5 mutations (38 prior to this study) by a further seven, including two missense, two splicing, one apparently in-frame deletion identified by MLPA and two stop mutations (Fig. 5). Four of these mutations affect the predicted NH2-terminal catalytic domain ending at residue 297; the p.Leu302AspfsX49 mutation (patient 5) maps just downstream, while the p.Arg550X (patient 3) and c.2376 + 5G > A (patient 7) mutations affect a scarcely involved kinase region. It may not be coincidental that the seizures of patient 3 have been controlled by drugs for 3 years and disappeared in patient 7, thus indicating a less severe evolution of the disease, while patient 5 (enrolled in the AS–AS-like group) is able to sit up alone and walk. These observations are in line with a recent study indicating that a less severe phenotype is associated with mutations in the second part of the protein [7]. It is also worth noting that the two described stop mutations probably undergo RNA nonsense-mediated decay and we proved this for p.Arg550X as no mutant RNA was found by RT-PCR. On the contrary, both of our de novo missense mutations are associated with a very severe clinical presentation, and the RT-PCR detection of the transcript for p.Ile72Thr makes it plausible to hypothesise that an aberrant protein may be more harmful than its absence. The finding of a small peak for the mutated allele by sequencing may be accounted for by the low sensitivity of this method to reveal the reduced amount of transcript. In our small series of CDKL5-mutated patients, we observed that the stop codon mutation carriers have a milder phenotype than those with missense or splicing mutations. Our patient 6 carries p.His127Arg, the first mosaic CDKL5 mutation described so far and the only missense mutation affecting the active serine–threonine kinase site and shows clear regression and severe hypotonia, but no microcephaly; furthermore, the severity of her phenotype parallels that of patient 2, who bears the missense mutation p.Ile72Thr, which involves a residue that may represent a small mutational hotspot affecting the catalytic kinase site. Indeed, the p.Ile72Thr mutation of patient 2 is the third affecting Ile72 to be discovered. A different change (p.Ile72Asn) was reported by Evans [21] and a silent change (p.Ile72Ile) was observed in a Chinese Rett cohort [4] and has so far been excluded as a polymorphism.
An examination of both our missense sequence changes using the splice-site prediction programme (ESE Finder release 2.0: http://rulai.cshl.edu/tools/ESE) predicts that these mutations do not significantly alter exonic splicing enhancer sites, thus suggesting that the aberrant transcripts can be translated into aberrant proteins. A severe phenotype has recently been reported in the case of two other missense mutations leading to mislocalised protein, which the authors suggested may be due to disruption caused by the amino acid substitution of nuclear import domains [5].
RT-PCR and sequencing allowed us to classify the splicing mutation of patient 1 as a truncating mutation, but the fact that the aberrant truncated transcript could be observed suggests that the shorter mutated allele does not undergo nonsense-mediated RNA decay. This mutation, that we already described [14], has been recently reported in another patient [6] who is defined as an atypical Rett. Our patient is similar in the most frequent features, but differs for seizure onset, autonomic features and sleep disturbance. The c.145 + 2 T > C mutation may thus pinpoint another mutation hotspot. In the case of the deletion identified by MLPA (patient 4), transcript analysis showed that aberrant transcripts in which the first two coding exons are deleted together with the initial ATG site also escape RNA decay. However, despite that a mutant transcript could be observed, biocomputer tools did not reveal any alternative ATG sites predicting a translatable protein. It seems that classifying mutations at DNA level may sometimes be insufficient to predict their consequences and address genotype–phenotype correlations.
In conclusion, bioinformatic analysis of CDKL5 mutations, coupled to RNA analysis on fresh blood or X inactivation profile-matched lymphoblastoid cell lines, constitute the necessary prerequisite to fully address the genotype–phenotype correlation in CDKL5 mutated female carriers.
References
Kalscheuer VM, Tao J, Donnelly A, Hollway G, Schwinger E, Kubart S, Menzel C, Hoeltzenbein M, Tommerup N, Eyre H, Harbord M, Haan E, Sutherland GR, Ropers HH, Gecz J (2003) Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet 72:1401–1411. doi:10.1086/375538
Grosso S, Brogna A, Bazzotti S, Renieri A, Morgese G, Balestri P (2007) Seizures and electroencephalographic findings in CDKL5 mutations: case report and review. Brain Dev 29(4):239–242. doi:10.1016/j.braindev.2006.09.001
Nectoux J, Heron D, Tallot M, Chelly J, Bienvenu T (2006) Maternal origin of a novel C-terminal truncation mutation in CDKL5 causing a severe atypical form of Rett syndrome. Clin Genet 70:29–33. doi:10.1111/j.1399-0004.2006.00629.x
Li M, Pan H, Bao XH, Zhang YZ, Wu XR (2007) MECP2 and CDKL5 gene mutation analysis in Chinese patients with Rett syndrome. J Hum Genet 52:38–47. doi:10.1007/s10038-006-0079-0
Rosas-Vargas H, Bahi-Buisson N, Philippe C, Nectoux J, Girard B, N'Guyen MA, Gitiaux C, Lazzaro L, Odent S, Jonveaux P, Chelly J, Bienvenu T (2008) Impairment of CDKL5 nuclear localization as a cause for severe infantile encephalopathy. J Med Genet 45:172–178. doi:10.1136/jmg.2007.053504
Bahi-Buisson N, Nectoux J, Rosas-Vargas H, Milh M, Boddaert N, Girard B, Cances C, Ville D, Afenjar A, Rio M, Héron D, N'Guyen Morel MA, Arzimanoglou A, Philippe C, Jonveaux P, Chelly J, Bienvenu T (2008) Key clinical features to identify girls with CDKL5 mutations. Brain 131:2647–2661. doi:10.1093/brain/awn197
Bahi-Buisson N, Kaminska A, Boddaert N, Rio M, Afenjar A, Gérard M, Giuliano F, Motte J, Héron D, Morel MA, Plouin P, Richelme C, des Portes V, Dulac O, Philippe C, Chiron C, Nabbout R, Bienvenu T (2008) The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia 49(6):1027–1037. doi:10.1111/j.1528-1167.2007.01520.x
Montini E, Andolfi G, Caruso A, Buchner G, Walpole SM, Mariani M, Consalez G, Trump D, Ballabio A, Franco B (1998) Identification and characterization of a novel serine threonine kinase gene from the Xp22 region. Genomics 51:427–433. doi:10.1006/geno.1998.5391
Lin C, Franco B, Rosner MR (2005) CDKL5/Stk9 kinase inactivation is associated with neuronal developmental disorders. Hum Mol Genet 24:3775–3786. doi:10.1093/hmg/ddi391
Rusconi L, Salvatoni L, Giudici L, Bertani I, Kilstrup-Nielsen C, Broccoli V, Landsberger N (2008) CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J Biol Chem 283(44):30101–30111 doi:10.1074/jbc.M804613200
Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R, Scala E, Longo I, Grosso S, Pescucci C, Ariani F, Hayek G, Balestri P, Bergo A, Badaracco G, Zappella M, Broccoli V, Renieri A, Kilstrup-Nielsen C, Landsberger N (2005) CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet 14:1935–1946. doi:10.1093/hmg/ddi198
Bertani I, Rusconi L, Bolognese F, Forlani G, Conca B, De Monte L, Badaracco G, Landsberger N, Kilstrup-Nielsen C (2006) Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J Biol Chem 281(42):32048–32056. doi:10.1074/jbc.M606325200
Weaving CJ, Ellaway J Gécz, Christodoulou J (2005) Rett syndrome: clinical review and genetic update. J Med Genet 42:1–7. doi:10.1136/jmg.2004.027730
Pintaudi M, Baglietto MG, Gaggero R, Parodi E, Pessagno A, Marchi M, Russo S, Veneselli E (2008) Clinical and electroencephalographic features in patients with CDKL5 mutations: two new Italian cases and review of literature. Epilepsy Behav 12:326–331. doi:10.1016/j.yebeh.2007.10.010
Elia M, Falco M, Ferri R, Spalletta A, Bottitta M, Calabrese G, Carotenuto M, Musumeci SA, Lo Giudice M, Fichera M (2008) CDKL5 mutations in boys with severe encephalopathy and early-onset intractable epilepsy. Neurol (Tokyo) 71:997–999
Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J (2006) Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A 140:413–418. doi:10.1002/ajmg.a.31074
Mnatzakanian GN, Lohi H, Munteanu I, Alfred SE, Yamada T, MacLeod PJM, Jones JR, Scherer SW, Schanen NC, Friez MJ, Vincent JB, Minassian BA (2004) A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat Genet 36(4):339–341. doi:10.1038/ng1327
Muralidhar B, Butler MG (1998) Methylation PCR analysis of Prader-Willi syndrome, Angelman syndrome, and control subjects. Am J Med Genet 16(80):263–265. doi:10.1002/(SICI) 1096-8628(19981116) 80:3<263::AID-AJMG16>3.0.CO;2-G
Scala E, Ariani F, Mari F, Caselli R, Pescucci C, Longo I, Meloni I, Giachino D, Bruttini M, Hayek G, Zappella M, Renieri A (2005) CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet 42:103–107. doi:10.1136/jmg.2004.026237
Allen RC, Zoghby HY, Mosely AB, Rosenblatt HM, Belmon JW (1992) Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 51:1229–1239
Evans JC, Hayley L, Colley JP, Ravn K, Nielsen JB, Kerr A, Williams E, Christodoulou J, Gecz J, Jardine PE, Wright MJ, Pilz DT, Lazarou L, Cooper DN, Sampson JR, Butler R, Whatley SD, Clarke AJ (2005) Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet 13:1113–1120. doi:10.1038/sj.ejhg.5201451
Tao J, Esch VH, Hagedorn-Greiwe M, Hoffman K, Moser B, Raynaud M, Sperner J, Fryns JP, Schwinger E, Gezc J, Ropers HH, Kalsheuer VM (2004) Mutations in the X-linked Cyclin-dependent Kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet 75:1149–1154. doi:10.1086/426460
Nishimura A, Takano T, Mizuguchi T, Saitsu H, Takeuchi Y, Matsumoto N (2008) CDKL5 distruption by t(X;18) in a girl with West syndrome. Clin Genet 74(3):288–290
Watson P, Black G, Ramsden S, Barrow M, Super M, Kerr B, Clayton-Smith J (2001) Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J Med Genet 38:224–228. doi:10.1136/jmg.38.4.224
Acknowledgements
We thank A.I.R. (Associazione Italiana Rett) for support of this study by a dedicated grant and for making available their daughters' samples and Galliera Genetic Bank for establishment of lymphoblastoid cell lines. This study was supported by the European grant E-Rare JTC 2007 (to S.R.) entitled European Network on Rett Syndrome.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
Supplementary Table 2
(XLS 17 KB)
Supplementary Fig. 6
Electropherogram of the splicing mutation affecting IVS16 carried by patient 7 and spliceview analysis, which predicts splicing consensus abolition (GIF 219 KB)
Rights and permissions
About this article
Cite this article
Russo, S., Marchi, M., Cogliati, F. et al. Novel mutations in the CDKL5 gene, predicted effects and associated phenotypes. Neurogenetics 10, 241–250 (2009). https://doi.org/10.1007/s10048-009-0177-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-009-0177-1