
Abstract.
The autosomal dominant cerebellar ataxias (ADCAs) are a clinically and genetically heterogeneous group of disorders. To date, at least 11 genes and 13 additional loci have been identified in ADCAs. Despite phenotypic differences, spinocerebellar ataxia 4 (SCA4) and Japanese 16q-linked ADCA type III map to the same region of 16q22.1. We report four Japanese families with pure cerebellar ataxia and a disease locus at 16q22.1. Our families yielded a peak lod score of 6.01 at marker D16S3141. To refine the candidate region, we carried out genetic linkage studies in four pedigrees with a high density set of DNA markers from chromosome 16q22.1. Our linkage data suggest that the disease locus for 16q-ADCA type III is within the 1.25-Mb interval delineated by markers 17msm and CTTT01. We screened for mutations in 36 genes within the critical region. Our critical region lies within the linkage interval reported for SCA4 and for Japanese 16q-ADCA type III. These data suggest that the ADCA that we have characterized is allelic with SCA4 and Japanese 16q-linked ADCA type III.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autosomal dominant cerebellar ataxias (ADCAs) are clinically and genetically heterogeneous. ADCAs are classified into three subgroups according to their clinical features [1]. ADCA type I is characterized by ophthalmoplegia, optic atrophy, dementia, and extrapyramidal features. ADCA type II is characterized by retinopathy and extrapyramidal features. ADCA type III is characterized by isolated late-onset cerebellar ataxia.
To date, 11 genes and 13 additional loci have been associated with ADCAs. The causative mutations in 9 of these 11 genes are expansions of a repetitive element. Expansions of polyglutamine tracts account for spinocerebellar ataxia 1 (SCA1) [2], SCA2 [3,4], SCA3 [5], SCA6 [6], SCA7 [7], and SCA17 [8]. Expansion of a CTG repeat within a non-coding RNA is responsible for SCA8 [9], expansion of the pentanucleotide ATTCT repeat within intron 9 of the SCA10 gene causes SCA10 [10], and a CAG repeat expansion within the promoter of PPP2R2B causes SCA12 [11]. Recently, missense mutations of PKCγ have been identified as a cause of SCA14 [12], and a mutation of FGF14 has been reported as a cause of SCA [13]. In addition to these identified disease genes, disease loci have been mapped for SCA4 [14], SCA5 [15], SCA11 [16], SCA13 [17], SCA15 [18], SCA16 [19], SCA18 [20], SCA19 [21], SCA20 [22], SCA21 [23], SCA22 [24], SCA24 [25], and SCA25 [26], although the responsible gene has not been identified yet.
SCA4 is an ADCA type I and is characterized by cerebellar ataxia with sensory axonal neuropathy. The SCA4 disease locus was originally mapped to 16q22.1 in a Utah family [14] and has subsequently been confirmed in SCA4 families from Germany [27].
Interestingly, the Japanese ADCA families that have been mapped to 16q22.1 (16q-ADCA type III) belong to ADCA type III because those patients have pure cerebellar ataxia [28]. On this basis, therefore, others have speculated that these Japanese patients have an allelic disorder of SCA4. Currently, the candidate disease locus of SCA4 encompasses an approximately 3.69-cM interval between D16S3019 and D16S512 and overlaps the 3.8-Mb disease locus between GGAA05 and D16S3095 that was mapped in the Japanese families [27,29].
Using four families living in southern Japan and segregating ADCA type III, we linked ADCA in these families to the SCA4 disease locus and narrowed the interval for the Japanese families with 16q-ADCA type III from 3.8 Mb to 1.25 Mb. Also, by direct sequencing of characterized and predicted coding exons within this interval, we excluded coding mutations within genes annotated in this interval.
Materials and methods
Human subjects and clinical assessment
Four ADCA type III families were studied (families M01, M02, M03, and K01). All four families originate from southern Japan. Expert neurologists examined all family members. We defined affected individuals as only those with cerebellar signs.
In total, we examined 30 family members, of whom 20 were affected. We list the ages of unaffected family members in Fig. 1. All patients were referred by their primary physician or neurologist and signed informed consents approved by the Institutional Review Board of Kagoshima University.

Pedigrees of the four families with autosomal dominant cerebellar ataxia (ADCA) type III. Filled symbols indicate affected individuals and open symbols indicate healthy subjects. Family members less than 60 years and without clinical symptoms are indicated by question marks. The ages of unaffected family members are listed under the linkage data. Oblique slashes indicate deceased family members. Squares represent men and circles women. The conserved disease haplotype in each family is boxed
Mutation analysis of known SCA genes
We isolated DNA from peripheral blood leukocytes of each patient. Prior to entry into our study, we screened each patient for all triplet repeat expansions associated with SCA (SCA1–3, 6–8, 12, 17, DRPLA) [2, 3, 4, 5, 6, 7, 8, 9, 11,30].
Linkage analysis
We performed linkage analyses of all family members using markers specific for identified SCA loci: chromosome 16q22.1 for SCA4 [14,27], chromosome 11p12-q12 for SCA5 [15], chromosome 22q13 for SCA10 [10], chromosome 15q14–21.3 for SCA11 [16], chromosome 19q13.3-q13.4 for SCA13 [17], chromosome 3p24.2–3pter for SCA15 [18,31], chromosome 8q23–24.1 for SCA16 [19], chromosome 7q22-q32 for SCA18 [20], chromosome 1p21-q21 for SCA19 [21], chromosome 7p21.3-p15.1 for SCA21 [23], and chromosome 1p21-q23 for SCA22 [24]. After screening these known loci by linkage analysis and identifying linkage to the SCA4 locus, we analyzed 18 markers including 3 new CA-repeat markers covering the 3.8-Mb 16q-ADCA type III region of the chromosome16q22.1 (TTCC01, D16S3086, GATA01, D16S421, CATG003, 17msm, D16S3107, D16S3085, 23msm, 25msm, D16S3025, TCTA01, CTTT01, D16S496, D16S3141, D16S3067, GT01, D16S3095). Marker information was obtained from the NCBI database and a previous report [29]. For the fine mapping, we designed 3 new CA-repeat markers (17msm, 23msm, 25msm). 17 msm F/R (5′-ATGACCCCCTGGTCACTATG-3′, 5′-CCATGTGCTTCAGGGAAGAT-3′); 23msm F/R (5′-CCAATCAAGTGTAGGGTGTGC-3′, 5′-ACCTAGGCCGGGTATGGTG-3′); 25msm F/R (5′-CAACATCCAAGCCCATCAGT-3′, 5′-CCCATGTGTGTTTCAGTCTTC-3′)
PCR amplifications were performed as follows: aliquots (50 ng) of genomic DNA were used as templates with fluorescently labeled primers, HotStarTaq Polymerase (Qiagen), reaction buffer, and dNTPs. After initial denaturation at 95°C for 15 min, amplification was performed for 40 cycles with denaturation at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 1 min, and a 30-min final extension step at 72°C. PCR products and the size standard (TAMRA-GS500) were analyzed using an ABI Prism 377 Genetic Analyzer. The genotypes were determined using the GeneScan Analysis (version3.1.2) and Genotyper (version2.5) programs. Two-point linkage analysis was performed with the MLINK program of the FASTLINK (v 5.2) software package [32]. Autosomal dominant inheritance was assumed in these families. Based on our epidemiological study (data not shown), we set the disease allele frequency at 0.00001. Because the penetrance of this disease is a function of age, we assumed that the penetrance was 0 for persons aged 45 years and younger, that it was 1 for persons 65 years and older, and that for persons between 45 and 65 years it was 0.05 (age−45), which is the equation of the distribution function. Lod scores were calculated assuming equal allele frequencies.
Mutation screening
We screened for mutations in 21 known genes and 15 predicted genes defined by the NCBI annotation (http://www.ncbi.nlm.nih.gov/RefSeq/key.html#query). We screened each gene by sequencing the coding exons in affected patients. For published cDNAs, we identified coding exons by aligning the genomic and cDNA sequences; for predicted genes in the NCBI database, we accepted the predicted exons comprising the open reading frame. Using the Primer v3 program, we designed primers to amplify exons and intronic splice junctions. By PCR we amplified the coding exons of each gene from 50 ng of patient genomic DNA using these primers and hot start PCR method as defined for amplification of polymorphic markers. Using the pre-sequencing kit (USB), we purified patient PCR products and sequenced them with dye terminator chemistry using an ABI377 automated sequencer (Applied Biosystems). We aligned the resulting sequences with the Sequencher sequence alignment program (Gene Codes). We numbered the nucleotides beginning with the adenine of the presumed initiating methionine and described mutations using standard nomenclature [33].
Results
Clinical features
The average age at onset was 56.7 years, and the existence of anticipation was unclear, because we could not obtain the accurate ages of onset for previous generations. Most of patients had a similar clinical phenotype: progressive ataxia, cerebellar signs, increased deep tendon reflexes, normal sensations, normal intelligence, and cerebellar atrophy on magnetic resonance imaging.
Mutation analysis and linkage of known SCA genes
We did not observe disease-associated triplet repeat expansions of SCA genes in any affected family members. Furthermore, by linkage analysis, we excluded association with the SCA disease loci for SCA5, SCA10, SCA11, SCA13, SCA15, SCA16, SCA18, SCA19, SCA21, and SCA22 (data not shown). However, we obtained a positive two-point lod score at D16S515 for SCA4/16q-ADCA type III. To verify this association, we screened 18 additional linkage markers located in the SCA4/16q-ADCA type III candidate region between D16S3043 and D16S3095. By two-point analysis, the highest lod score was obtained at D16S3141 (Z=6.01 at theta=0) and the lod scores of 8 markers exceeded 3.0 (Table 1). On haplotype analysis, all affected members in families M01, M02, and M03 showed the same haplotype from the marker TTCC01 to D16S3095. Genetic cross-over was detected between TCTA01 and CTTT01 in family K01.
All four of our families had a common disease-associated haplotype 6–2-3–2-2–2 for markers D16S3107, D16S3085, 23msm, 25msm, D16S3025, and TCTA01, respectively (Fig. 2). These data suggest a single founder and place the disease locus between markers 17msm and CTTT01, a distance of 1.25 Mb. Consanguineous marriage was present in family M03 and consistent with this, IV-12 had a homozygous disease-associated haplotype in the critical region.

Haplotype analysis of 16q22.1 in four families. The disease-associated haplotypes are shown. Common alleles are indicated by dotted lines
Mutation screening of the genes within the critical region
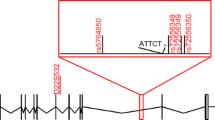
The physical map of the region of 16q22 on which our analyses focused is shown in Fig. 3. The critical region we identified within this locus contains 21 known genes and 15 predicted genes from the NCBI annotation. The DNA sequencing of the coding exons and 50 bases of the flanking introns were performed in at least two patients from each family. We found 9 sequence variants (two insertion/deletion mutations and 7 single nucleotide changes) in the coding exons of 7 of 36 genes (Table 2). The heterozygous mutation 822insG in ZFP90 causes a frameshift mutation leading to premature termination, but it does not co-segregate with the disease allele in these families and was also detected in control DNA. THAP11 contains CAG repeats interrupted by CAA in the coding exon, which encodes 29 polyglutamines. We found one deletion of 29 polyglutamines in our patients, but this variant did not co-segregate with the disease. We confirmed the un-expanded PCR products of THAP11 in our patients with a homozygous disease-associated haplotype to rule out the possibility of a large CAG expansion. Four novel sequence variants were detected in 4 genes (Table 2); however, these variants did not co-segregate with the disease in our families.

Physical map of 16q22. This map shows the critical region of SCA4/16q-ADCA type III and is based on the human genome sequence NT_010498.14. This figure shows the distance of microsatellite markers. The three bars indicate the critical regions in the study [14, 27, 28,29]. There are 36 candidate genes in our critical region
Discussion
The SCA4 locus was first mapped to 16q22.1 in a large Utah family in 1996 [14] and was refined to a 3.69-cM region between D16S3019 and D16S512 in German and Utah families in 2003 [27]. Japanese ADCA families also mapped within the SCA4 candidate region, and the disease was reported as “16q-linked ADCA type III” [28,29]. The clinical phenotype of the Japanese patients is clearly distinguishable from SCA4, and it is unclear whether or not they are allelic disorders. However, our critical region resides within the newly reported intervals of SCA4 and 16q-ADCA type III; this again suggests that SCA4 and 16q-ADCA type III could be allelic disorders.
We report four new Japanese families with ADCA with disease linkage to chromosome 16q22.1. All family members are from the southern part of Japan. According to the clinical classification of ADCAs, our families belong to ADCA type III, and now we have shown that they also have 16q-ADCA type III.
Comparing the molecular data from our families with that of the reported Japanese families with 16q-ADCA type III, we observed that both showed the same-sized PCR products for five DNA markers [GT01 (189 bp), CTTT01 (242 bp), TCTA01 (219 bp), CATG003 (194 bp), GATA01 (158 bp)] and different-sized PCR product for TTCC01 [26] Based on the previous report, DNA samples were collected from a wider area of Japan. The similarities of the clinical phenotypes between our families and the previously reported Japanese 16q-ADCA type III families and the occurrence of the same haplotype in five adjacent markers suggest that most Japanese families segregating 16q-ADCA type III originated from a single founder. Therefore, we utilized the founder haplotype for our mapping and this narrowed the candidate region (Figs. 2 and 3). Finally, we reduced the critical interval to 1.25 Mb. Although there were at least 96 candidate genes for the minimum interval from previous studies [29], our linkage study reduced the candidate genes to 36.
Based on our fine mapping data, we screened the coding exons of the 36 candidate genes (21 known genes and 15 predicted genes). We identified 5 single nucleotide polymorphisms and 4 novel variants. Assuming a common founder mutation, we also excluded deletions of any exons by PCR amplification of each exon from a patient homozygous for the disease-associated allele (data not shown); our method would have detected any homozygous deletion greater than 20 base pairs. Our observations suggest that the disease-causing mutation could arise from a genomic rearrangement or be in the promotor region, intron, or a non-coding exon of a gene that we screened or in an unidentified gene. In summary, this report provides new linkage information and thereby will facilitate the identification of the genetic basis of SCA4/16q-ADCA type III.
References
Harding AE (1993) Clinical features and classification of inherited ataxias. Adv Neurol 61:1–14
Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4:221–226
Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, Weber C, Mandel JL, Cancel G, Abbas N, Durr A, Didierjean O, Stevanin G, Agid Y, Brice A (1996) Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 14:285–291
Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A, Tashiro K, Ishida Y, Ikeuchi T, Koide R, Saito M, Sato A, Tanaka T, Hanyu S, Takiyama Y, Nishizawa M, Shimizu N, Nomura Y, Segawa M, Iwabuchi K, Eguchi I, Tanaka H, Takahashi H, Tsuji S (1996) Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 14:277–284
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I, et al (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8:221–228
Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC (1997) Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet 15:62–69
David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E, Drabkin H, Gemmill R, Giunti P, Benomar A, Wood N, Ruberg M, Agid Y, Mandel JL, Brice A (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17:65–70
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10:1441–1448
Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, Ranum LP (1999) An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet 21:379–384
Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, Achari M, Pulst SM, Alonso E, Noebels JL, Nelson DL, Zoghbi HY, Ashizawa T (2000) Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet 26:191–194
Holmes SE, O’Hearn EE, McInnis MG, Gorelick-Feldman DA, Kleiderlein JJ, Callahan C, Kwak NG, Ingersoll-Ashworth RG, Sherr M, Sumner AJ, Sharp AH, Ananth U, Seltzer WK, Boss MA, Vieria-Saecker AM, Epplen JT, Riess O, Ross CA, Margolis RL (1999) Expansion of a novel CAG trinucleotide repeat in the 5’ region of PPP2R2B is associated with SCA12. Nat Genet 23:391–392
Chen DH, Brkanac Z, Verlinde CL, Tan XJ, Bylenok L, Nochlin D, Matsushita M, Lipe H, Wolff J, Fernandez M, Cimino PJ, Bird TD, Raskind WH (2003) Missense mutations in the regulatory domain of PKC gamma: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet 72:839–849
Swieten JC van, Brusse E, Graaf BM de, Krieger E, Graaf R van de, Koning I de, Maat-Kievit A, Leegwater P, Dooijes D, Oostra BA, Heutink P (2003) A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia. Am J Hum Genet 72:191–199
Flanigan K, Gardner K, Alderson K, Galster B, Otterud B, Leppert MF, Kaplan C, Ptacek LJ (1996) Autosomal dominant spinocerebellar ataxia with sensory axonal neuropathy (SCA4): clinical description and genetic localization to chromosome 16q22.1. Am J Hum Genet 59:392–399
Ranum LP, Schut LJ, Lundgren JK, Orr HT, Livingston DM (1994) Spinocerebellar ataxia type 5 in a family descended from the grandparents of President Lincoln maps to chromosome 11. Nat Genet 8:280–284
Worth PF, Giunti P, Gardner-Thorpe C, Dixon PH, Davis MB, Wood NW (1999) Autosomal dominant cerebellar ataxia type III: linkage in a large British family to a 7.6-cM region on chromosome 15q14–21.3. Am J Hum Genet 65:420–426
Herman-Bert A, Stevanin G, Netter JC, Rascol O, Brassat D, Calvas P, Camuzat A, Yuan Q, Schalling M, Durr A, Brice A (2000) Mapping of spinocerebellar ataxia 13 to chromosome 19q13.3-q13.4 in a family with autosomal dominant cerebellar ataxia and mental retardation. Am J Hum Genet 67:229–235
Knight MA, Kennerson ML, Anney RJ, Matsuura T, Nicholson GA, Salimi-Tari P, Gardner RJ, Storey E, Forrest SM (2003) Spinocerebellar ataxia type 15 (SCA15) maps to 3p24.2–3pter: exclusion of the ITPR1 gene, the human orthologue of an ataxic mouse mutant. Neurobiol Dis 13:147–157
Miyoshi Y, Yamada T, Tanimura M, Taniwaki T, Arakawa K, Ohyagi Y, Furuya H, Yamamoto K, Sakai K, Sasazuki T, Kira J (2001) A novel autosomal dominant spinocerebellar ataxia (SCA16) linked to chromosome 8q22.1–24.1. Neurology 57:96–100
Brkanac Z, Fernandez M, Matsushita M, Lipe H, Wolff J, Bird TD, Raskind WH (2002) Autosomal dominant sensory/motor neuropathy with ataxia (SMNA): linkage to chromosome 7q22-q32. Am J Med Genet 114:450–457
Verbeek DS, Schelhaas JH, Ippel EF, Beemer FA, Pearson PL, Sinke RJ (2002) Identification of a novel SCA locus ( SCA19) in a Dutch autosomal dominant cerebellar ataxia family on chromosome region 1p21-q21. Hum Genet 111:388–393
Knight MA, Gardner RJ, Bahlo M, Matsuura T, Dixon JA, Forrest SM, Storey E (2004) Dominantly inherited ataxia and dysphonia with dentate calcification: spinocerebellar ataxia type 20. Brain 127:1172–1181
Vuillaume I, Devos D, Schraen-Maschke S, Dina C, Lemainque A, Vasseur F, Bocquillon G, Devos P, Kocinski C, Marzys C, Destee A, Sablonniere B (2002) A new locus for spinocerebellar ataxia (SCA21) maps to chromosome 7p21.3-p15.1. Ann Neurol 52:666–670
Chung MY, Lu YC, Cheng NC, Soong BW (2003) A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21-q23. Brain 126:1293–1299
Swartz BE, Burmeister M, Somers JT, Rottach KG, Bespalova IN, Leigh RJ (2002) A form of inherited cerebellar ataxia with saccadic intrusions, increased saccadic speed, sensory neuropathy, and myoclonus. Ann N Y Acad Sci 956:441–444
Stevanin G, Bouslam N, Thobois S, Azzedine H, Ravaux L, Boland A, Schalling M, Broussolle E, Durr A, Brice A (2004) Spinocerebellar ataxia with sensory neuropathy (SCA25) maps to chromosome 2p. Ann Neurol 55:97–104
Hellenbroich Y, Bubel S, Pawlack H, Opitz S, Vieregge P, Schwinger E, Zuhlke C (2003) Refinement of the spinocerebellar ataxia type 4 locus in a large German family and exclusion of CAG repeat expansions in this region. J Neurol 250:668–671
Nagaoka U, Takashima M, Ishikawa K, Yoshizawa K, Yoshizawa T, Ishikawa M, Yamawaki T, Shoji S, Mizusawa H (2000) A gene on SCA4 locus causes dominantly inherited pure cerebellar ataxia. Neurology 54:1971–1975
Li M, Ishikawa K, Toru S, Tomimitsu H, Takashima M, Goto J, Takiyama Y, Sasaki H, Imoto I, Inazawa J, Toda T, Kanazawa I, Mizusawa H (2003) Physical map and haplotype analysis of 16q-linked autosomal dominant cerebellar ataxia (ADCA) type III in Japan. J Hum Genet 48:111–118
Nagafuchi S, Yanagisawa H, Ohsaki E, Shirayama T, Tadokoro K, Inoue T, Yamada M (1994) Structure and expression of the gene responsible for the triplet repeat disorder, dentatorubral and pallidoluysian atrophy (DRPLA). Nat Genet 8:177–182
Storey E, Gardner RJ, Knight MA, Kennerson ML, Tuck RR, Forrest SM, Nicholson GA (2001) A new autosomal dominant pure cerebellar ataxia. Neurology 57:1913–1915
Lathrop GM, Lalouel JM (1984) Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 36:460–465
Dunnen JT den, Antonarakis SE (2000) Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 15:7–12
Acknowledgements.
We thank the families described for co-operation and Dr. C.F. Boerkoel of Baylor College of Medicine and Dr. A.R. Ng of Kagoshima University Faculty of Medicine for critical review. We also thank Ms. S. Taniguchi of Kagoshima University for her excellent technical assistance. This study was supported in part by grants from the Cerebellar Ataxia, Neurodegenerative Diseases and Inherited Neuropathy of the Ministry of Health and Welfare of Japan to M.O., M.N., and K.A., from the Kanae Foundation and the Nakabayashi Trust for ALS research to H.T.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hirano, R., Takashima, H., Okubo, R. et al. Fine mapping of 16q-linked autosomal dominant cerebellar ataxia type III in Japanese families. Neurogenetics 5, 215–221 (2004). https://doi.org/10.1007/s10048-004-0194-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-004-0194-z