
Abstract
Concentrated growth factor (CGF) is an autologous leukocyte-rich and platelet-rich fibrin (L-PRF) biomaterial termed “second-generation platelet concentrate”. CGF contains autologous osteoinductive platelet growth factors and an osteoconductive fibrin matrix. The purpose of this study was to assess the ability of CGF combined with bone marrow stromal cells (BMSCs) to heal critical-size rat calvaria defects in vivo and to modulate the proliferation and osteogenic differentiation of mesenchymal stem cells (MSCs) in vitro. In the in-vivo study, the CGF group regenerated bone better than the control group, and combined therapy with CGF and BMSCs almost completely repaired critical-size bone defects within 12 weeks after surgery. In the in-vitro study, the CGF extract, at concentrations between 1 and 10 %, promoted proliferation, osteogenic maturation, and mineralization of hTERT-E6/E7 human MSCs in a dose-dependent manner but had an inhibitory effect at higher concentrations. In conclusion, a CGF extract promoted the proliferation, osteogenic maturation, and mineralization of mesenchymal stem cells in vitro, and combination therapy with CGF and BMSCs resulted in excellent healing of critical-size bone defects in vivo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Interest has recently been focused on stem or progenitor cell therapy to replace or repair damaged or lost tissue (because of disease or injury) by tissue engineering. Cell therapy for bone regeneration requires an osteoconductive scaffold and growth factors for osteoinduction. Platelets contain a variety of autologous growth factors, including platelet-derived growth factors (PDGF), transforming growth factors β (TGF-β), vascular endothelial growth factors (VEGF), and insulin-like growth factors (IGF), which are of crucial importance in bone healing. Platelets also secrete fibrinogen, and the extensive cross-linking of fibrin α-chains during clot formation strongly enhances the mesenchymal cell migration essential for tissue regeneration [1–5].
In 1998, Marx et al. [6] found that platelet-rich plasma (PRP), a concentrated preparation of platelets now termed “first-generation platelet concentrate”, had a positive effect on bone regeneration and wound healing, for which it functioned as a source of autologous growth factors. Since then, PRP has been widely used in many fields including dentistry, dermatology, orthopedics, cosmetics, and cardiothoracic surgery [7]. In general, this large body of PRP studies demonstrated that PRP stimulates the proliferation and differentiation of fibroblasts, osteoblasts, chondrocytes, and mesenchymal stem cells, although some contradictory results have been reported [8–10], probably because of the diversity of procedures used to isolate PRP.
Concentrated growth factor (CGF) is an autologous leukocyte-rich and platelet-rich fibrin (L-PRF) biomaterial termed “second-generation platelet concentrate” [11]. (Unlike PRP, CGF is obtained by single centrifugation of venous blood by use of a specially programmed centrifuge [12]). CGF contains autologous osteoinductive growth factors derived from platelets and an osteoconductive fibrin matrix. One of the main differences between CGF and most PRP preparations is that CGF production does not require addition of other reagents, i.e. it does not use anticoagulant during blood harvesting or heterozoic thrombin and calcium chloride for platelet activation and fibrin polymerization. Instead of using these reagents, CGF itself slowly polymerizes during centrifugation in a manner similar to natural polymerization in vivo. Such polymerization is crucial for proper three-dimensional (3D) organization of a fibrin network. A CGF-derived fibrin structure is favorable for cytokine enmeshment and cellular migration (because of the 3D organization of a fibrin network) and slowly releases platelet growth factors for at least 7–10 days [13, 14]. These combined observations suggest that combination therapy of CGF with bone marrow stromal cells (BMSCs) may provide all the necessary features (osteoproductive cells: BMSCs, osteoinductive scaffold: fibrin matrix, osteoconductive growth factors: platelet growth factors) for induction of bone growth and regeneration.
The purpose of this study was to determine the effect of the second-generation platelet concentrate, CGF, on human mesenchymal stem cells (hTERT-E6/E7) [15] in vitro and the feasibility of combined therapy of CGF with BMSCs for bone regeneration in a critical-size rat calvaria-defect model in vivo. Our study revealed that CGF promoted the proliferation, osteogenic maturation, and mineralization of mesenchymal stem cells in vitro, and combination therapy of CGF with BMSCs enabled excellent healing of a critical-size bone defect in vivo.
Materials and methods
CGF preparation
This research was approved by the institute’s committee on human research and the protocol was found acceptable by them. The experimental protocol was approved (approval number 09140(758-4)-5, date of approval July 28, 2010) by the Institutional Review Board of Osaka University. For the in-vitro experiments, CGF was prepared from human venous blood obtained from three healthy male volunteers (age range 26–37 years). All subjects enrolled in this research gave informed consent. Venous blood was taken, without anticoagulants, directly into a sterile 10-ml tube and was immediately centrifuged in a special centrifuge device (Medifuge®; Slifadent srl, Sofia, Italy) for 13 min. This centrifuge device used a program with the characteristics: 2,700 rpm 2 min, 2,400 rpm 4 min, 2,700 rpm 4 min, and 3,000 rpm 3 min.
For the in-vivo experiments, donor rats were anesthetized by intramuscular injection of a mixture of midazolam (4 mg/kg), medetomidine (1 mg/kg), and butorphanol (5 mg/kg). Blood was collected, by cardiac puncture, without anticoagulants, into a sterile 10-ml tube and was immediately centrifuged by use of a Medifuge® for 13 min. CGF from rats was prepared in the same way as that from humans.
Preparation of a CGF extract
Immediately after CGF preparation, CGF was transferred into a syringe and was gently compressed in the syringe so that it divided into a solid fibrin membrane and a liquid fraction (CGF extract) (Fig. 1).

Preparation of a CGF extract. a CGF was transferred into a syringe and was gently compressed in the syringe so that a liquid fraction (the CGF extract is indicated with white arrows) was separated from a solid fibrin membrane. b CGF c solid fibrin membrane (upper) and CGF extract (lower)
Cell culture
Cells of a human mesenchymal stem cell line, hTERT-E6/E7, were provided by Dr Junya Toguchida (Kyoto University, Japan). The cells were grown in α-minimum essential medium (α-MEM; Invitrogen, Carlsbad, CA, USA) supplemented with 1 % antibiotics (penicillin–streptomycin; Invitrogen) and 10 % fetal bovine serum (FBS; Hyclone, Road Logan, UT, USA) (growth medium) at 37 °C under a humidified atmosphere of 5 % CO2. Before each assay, the hTERT-E6/E7 cells were plated and pretreated in pretreatment medium (α-MEM containing 1 % FBS and 1 % antibiotics) for 12 h, and, after attachment, the medium was changed to differentiation medium (α-MEM containing 1 % FBS, 1 % antibiotics, 50 μM ascorbic acid (Sigma–Aldrich, St Louis, MO, USA), 10 mM β-glycerophosphate (Sigma–Aldrich), and 100 nM dexamethasone (Sigma–Aldrich)). The medium was changed every three days.
In the in-vitro proliferation assay and the differentiation assay, we used the human mesenchymal stem cell line, hTERT-E6/E7 because we assumed that using established cell line might produce stable experimental results, even though we needed to use freshly prepared CGFs from different volunteers in each experiment.
Proliferation assay
hTERT-E6/E7 cells were seeded in 6-well plates at a density of 5.0 × 103 cells/cm2 in pretreatment medium. After attachment, the pretreatment medium was changed to differentiation medium containing 0, 1, 3, 5, 10, or 20 % CGF extract or serum for 24 h. The number of cells was counted after detachment of the cells by use of 0.25 % trypsin EDTA (Invitrogen). All experiments were performed three times, with duplicates.
Alkaline phosphatase (ALP) staining
hTERT-E6/E7 cells were seeded in 24-well plates at a density of 2.0 × 104 cells/cm2. After incubation for 12 h in pretreatment medium, the medium was changed to differentiation medium containing 0, 1, 3, 5, 10, or 20 % CGF extract or serum for 4 days. For ALP staining, the cells were washed with phosphate-buffered saline (PBS; Sigma–Aldrich) and fixed for 5 min with 10 % formalin at room temperature. After fixing, the cells were incubated with the BCIP/NBT color development substrate (Promega, Madison, WI, USA) for 1 h at 37 °C. After staining, plates were digitally photographed and the acquired images were analyzed.
ALP activity
To measure ALP activity, cells were washed twice with PBS then lysed in mammalian protein extraction reagent (M-PER; Pierce, Rockford, IL, USA) in accordance with the manufacturer’s procedure. ALP activity was measured by use of LabAssay ALP (Wako Pure Chemicals Industries, Osaka, Japan) with p-nitrophenylphosphate as substrate. To normalize enzyme activity, the protein content of the solution was measured by use of a bicinchoninic acid (BCA) protein assay kit (Pierce).
Alizarin red staining
hTERT-E6/E7 cells were seeded in 24-well plates at a density of 2.0 × 104 cells/cm2. After incubation for 12 h in pretreatment medium, the medium was changed to differentiation medium containing 0, 1, 3, 5, 10, or 20 % CGF extract or serum for 21 days. The cells were then washed twice with PBS, fixed in 10 % formalin for 10 min, and then stained with alizarin red S (Sigma–Aldrich) at pH 6.3 for 1 h. After discarding the alizarin red S solution and washing the cells three times with distilled water, the bound alizarin red was dissolved in 200 μl 100 mM hexadecylpyridinium chloride (Sigma–Aldrich) and the absorbance of the supernatant was measured at 570 nm.
Von Kossa staining
hTERT-E6/E7 cells were seeded in 24-well plates at a density of 2.0 × 104 cells/cm2. After incubation for 12 h in pretreatment medium, the medium was changed to differentiation medium containing 0, 1, 3, 5, 10, or 20 % CGF extract or serum for 21 days. The cells were washed twice with PBS, fixed in 10 % formalin for 10 min, then 2 ml freshly prepared 1 % silver nitrate was added to each well and the wells were incubated under UV light for 30 min. The plates were washed with distilled water, fixed using 5 % sodium thiosulfate for 5 min, then washed thoroughly with distilled water to terminate the reaction. Macro photographs of mineral deposits were then recorded.
Quantitative real-time PCR
Total RNA was isolated from cells by use of the RNeasy mini Kit (Qiagen, Valencia, CA, USA), in accordance with the manufacturer’s instructions. cDNA was synthesized by use of the SuperScript III First-Strand synthesis system (Invitrogen). This cDNA was then analyzed by real-time PCR analysis by use of the 7900HT fast real-time PCR system (Applied Biosystems, Tokyo, Japan). The SYBR green assay with SYBR Premix EX taq (Tli RnaseH plus; TaKaRa Bio, Otsu, Japan) was used for amplification of all target transcripts. Expression values were normalized to GAPDH. The sequences of the specific primers used are shown in Table 1.
Measurement of platelet growth factor (PDGF-BB, TGF-β1, TNF-α, IL-1β) levels in CGF CGF, PRP (2,400 rpm 10 min, 3,600 rpm 15 min), and serum were assayed for typical growth factors using commercially available Quantikine ELISA kits (R&D Systems, Minneapolis, MN, USA). We incubated CGF in centrifuge tubes containing 1,000 μl PBS and stored for 13 days at 37 °C. The PBS in the tubes was replaced every 2 days and collected for ELISA.
BMSC isolation and culture
Bone marrow cells were obtained by flushing the bone marrow cavity of rat femoral and tibial bones with 5 ml saline. For BMSC isolation and expansion, the flushed bone marrow fluid was first passed through 90-μm pore strainers for isolation of bone spicules. Next, filtrates containing bone marrow cells were plated in a plastic culture dish in growth medium that had been screened for optimum BMSC growth. After incubation for 24 h, non-adherent cells were removed by means of 2 or 3 PBS washing steps. After 2–3 weeks of culture in this growth medium, the adherent cells were used as BMSCs for in-vivo assays.
Preparation of CGF with BMSCs
BMSCs (5.0 × 105) suspended in 20 μl PBS were injected into CGF (average weight 39.1 ± 1.99 mg) by use of a 1-ml syringe with a 26-gauge needle.
The rat calvaria critical-size bone defect model
Twenty-seven male Sprague–Dawley (SD) albino rats (10–12 weeks old, average weight 300–350 g) were randomly divided into three groups:
-
1.
unfilled defect (control);
-
2.
CGF alone; and
-
3.
CGF + BMSCs.
All animals were housed in cages with free access to food pellets and water. The experimental protocol was approved (Approval Number 21-063-0) by the Animal Experiment Committee of Osaka University, and the experiments were carried out in accordance with the Osaka University guidelines for the care and use of laboratory animals. All efforts were made to minimize suffering. General anesthesia was induced by intramuscular injection of a mixture of midazolam, medetomidine, and butorphanol. An incision was made midline from the supraorbital glabella to the occiput. This incision was continued through the periosteum, which was elevated from the underlying bone and retracted laterally. Two calvarial defects, 5 mm in diameter, were created symmetrically on the bilateral sides of the midline in each rat by use of a trephine bur without dura perforation. A sterile saline solution was used to keep membranes moist and to thoroughly remove bone debris. The CGF ± BMSCs were applied directly into the defect sites. The skin was closed with 5–0 nylon.
Microfocus computed tomography
Formation of new bone in each calvaria sample was evaluated by use of a microfocus computed tomography (micro-CT) system (SMX-100CT-SV; Shimadzu, Kyoto, Japan). Each sample was scanned at 50-μm intervals at 30 kV and 175 μA. After scanning, 3D CT images were reconstructed and the calcified volume of newly formed bone in the calvarial defect was measured by use of Tri/3D Bon software (Ratoc System Engineering, Tokyo, Japan).
Histological analysis
Samples were fixed in 10 % neutral formalin and were decalcified with ethylenediaminetetraacetic acid (pH 7.4). After decalcification, the samples were dehydrated in a graded ethanol series, cut along the coronal plane at the midline of the defect, and embedded in paraffin. Sections (3 μm thick) were each mounted on to individual slides and stained with H&E for observation under a light and polarized light microscope (Eclipse 90i; Nikon, Tokyo, Japan).
Statistical analysis
All data are expressed as mean ± SD and a minimum of three independent experiments were performed for each assay. A two-sided unpaired Student’s t test or analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons were used for statistical analysis of differences between groups. A statistical difference between experimental groups was regarded as significant when the p value was <0.05.
Results
Quantitative real-time PCR analysis of the effect of CGF extracts on expression of osteoblast-related genes
hTERT-E6/E7 cells were treated with CGF extract at concentrations of 1, 3, 5, 10, or 20 % in the culture medium for 3 days and CGF effects on osteoblastic gene expression were assessed by use of quantitative real-time PCR. Gene expression of ALP and osteopontin (OPN) was significantly increased by treatment with CGF extract (5–20 and 5–10 %, respectively) compared with treatment with the same concentration of serum (Fig. 2). Gene expression of Type 1 collagen alpha 1 (COL1A1) was also increased by treatment with 3–5 % CGF extract, but a high concentration (10–20 %) of the CGF extract or of serum reduced this gene expression compared with the 0 % control (differentiation medium only) group (Fig. 2). Gene expression of runt-related transcription factor 2 (RUNX2) and of osterix (OSX), the key regulators of osteogenesis, were not significantly changed by treatment with the CGF extract (Fig. 2).

Quantitative real-time PCR analysis of the effect of CGF extract on expression of osteoblast-related genes. Cells of the human mesenchymal stem cell line hTERT-E6/E7 were treated with different concentrations of a concentrated growth factors (CGF) extract or serum control for 3 days and mRNA expression of the indicated genes was evaluated by use of quantitative RT-PCR. Expression of each gene was normalized to GAPDH expression. All data are mean ± SD from three independent experiments (each experiment used CGF from a different volunteer), each performed in duplicate. (*P < 0.05 compared with the serum group of the same concentration.)
Effect of CGF extracts on proliferation of hTERT-E6/E7 cells
A previous study showed that a first-generation plasma concentrate (Ex.PRP) has a positive dose-dependent effect on cell proliferation [16]. However, other studies have reported the contradictory result that low concentrations of Ex.PRP (a few times greater than physiological levels) are more efficient at enhancing cell proliferation than very high concentrations [17–19]. In this study, the effect of a CGF extract on human mesenchymal stem cell proliferation was assessed by use of a cell count assay and hTERT-E6/E7 cells. In this study, the effect of a CGF extract or serum at concentrations of 1, 3, 5, 10, or 20 % in the culture medium was tested. hTERT-E6/E7 cell proliferation was promoted, in a dose-dependent fashion, by treatment with a CGF extract compared with the effect of serum addition. A significant difference between the effect of CGF and serum on cell proliferation was observed at all concentrations except 20 %. In particular, CGF extract at concentrations of 5 and 10 % markedly increased hTERT-E6/E7 proliferation (Fig. 3a).

Effect of a CGF extract on growth and osteoblastic differentiation of hTERT-E6/E7 mesenchymal cells. a Cell proliferation. hTERT-E6/E7 cells (5 × 103/well) were treated with different concentrations of a CGF extract or serum control for 24 h after which cell number was counted. b Alkaline phosphatase (ALP) staining and enzymatic activity. hTERT-E6/E7 cells were treated with different concentrations of a CGF extract or control serum for 4 days and were then subjected to ALP staining and activity assays. ALP activity was normalized to the protein content of each sample. c, d Mineralization. hTERT-E6/E7 cells were treated with different concentrations of a CGF extract or control serum for 21 days and were then stained with alizarin red S solution. The Ca content in the matrix of the cells was quantified by measuring the absorbance at 570 nm of the extract of the calcium bound dye. hTERT-E6/E7 cells were treated with different concentrations of a CGF extract or control serum for 21 days and were then stained with von Kossa solution. All data are mean ± SD from three independent experiments (each experiment used CGF from a different volunteer) performed in duplicate. (*P < 0.05 compared with the serum group of the same concentration.)
Effect of a CGF extract on the ALP staining and enzymatic activity of hTERT-E6/E7 cells
We next investigated the effect of a CGF extract on osteoblastic differentiation of hTERT-E6/E7 cells by ALP staining and ALP enzyme activity measurement after 4 days treatment with CGF or normal serum. CGF extract treatment increased ALP activity at all concentrations tested compared with the 0 % control (differentiation medium only) group. In addition when compared with equivalent serum dose groups, the CGF-induced increase in ALP activity was significantly different at CGF concentrations of 3–20 % (Fig. 3b).
Effect of CGF extract on mineralization of hTERT-E6/E7 cells
To further confirm the effect of CGF on osteoblastic differentiation, the effect of CGF on the mineralization of hTERT-E6/E7 cells was tested by alizarin red (Fig. 3c) and von Kossa staining (Fig. 3d). hTERT-E6/E7 cells were cultured with differentiation medium containing one of six different doses of a CGF extract or serum for 21 days. Alizarin red and von Kossa staining of these cells showed that CGF at concentrations between 3 and 10 % promoted calcification of the extracellular matrix (ECM) whereas calcification was not clearly observed in cells treated with CGF or serum at a concentration of 20 %. The extent of this ECM calcification was quantified by measurement of the absorbance of the alizarin red S solution. A CGF extract at a concentration between 3 and 10 % significantly increased calcification of the ECM compared with each equivalent concentration of serum.
Typical growth factors and inflammatory cytokines in CGF
We measured typical growth factors and inflammatory cytokines, including PDGF-BB, TGF-β1, TNF-α, and IL-1β, that were secreted by platelets. We found the CGF extract contained these cytokines at concentrations similar to or higher than those in serum. PRP contained high levels of inflammatory cytokines that might inhibit bone formation yet also contained high concentrations of cytokines that might promote bone formation. In a sustained-release test, we found CGF slowly released these cytokines for 9–13 days (Fig. 4).

Measurement of typical platelet cytokines. CGF, PRP, and serum were assayed for PDGF-BB, TGF-β1, TNF-α, and IL-1β by ELISA. In a sustained-release test we incubated CGF for 13 days at 37 °C in centrifuge tubes containing 1,000 μl PBS. The PBS in the tubes was replaced every 2 days and collected for ELISA. CGF slowly released these cytokines. a PDGF-BB, b TGF-β, c TNF-α, d IL-1β. ND not detected
Treatment of a rat calvaria critical-size bone defect with CGF and BMSCs: Micro-CT analysis
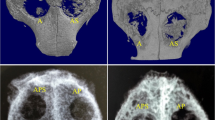
We next investigated the effect of CGF with or without BMSCs on bone regeneration using a representative critical-size bone defect model, the rat calvaria defect model. Representative 3D-reconstructed images of empty defect, CGF, and CGF + BMSC groups, assessed by micro-CT at 4, 8, and 12 weeks, are shown in Fig. 5a. An increase in bone formation was observed for all groups over the 12-week period. However, the CGF group and the CGF + BMSC group seemed to regenerate bone better than the control (empty defect) group.

Treatment of rat calvaria critical-size bone defects with CGF and BMSCs: micro-CT analysis. Bilateral, full thickness, critical-size bone defects (5.0 mm in diameter) were created in the parietal bones of Sprague–Dawley rats. These defects were treated by filling with CGF or CGF + bone marrow stromal cells (BMSCs), or were left unfilled as empty defect controls. a Representative microfocus computed tomography (micro-CT) images of cranial samples of the three groups 4, 8, and 12 weeks (W) after operation. Bar 5 mm. b Calculation of the volume of newly formed bone in calvaria defect sites in the three groups 4, 8, and 12 weeks after operation. All data are mean ± SD from six samples. (*P < 0.05)
In the control group, bone regeneration was observed only at the periphery of the defect over the 12-week period. In the CGF group, new bone formation started at the peripheral area of the defect and then proceeded toward the center of the defect over time. However, in the CGF + BMSC group, new bone formation was observed not only at the periphery of the bone hole but also in the central area of the defect. This new, central bone included islands of new bone that had no contact with the cut edge of the host bone and that were observed starting from 4 weeks after operation. This group had almost completely repaired critical-size bone defects within 12 weeks after operation.
The total volume of mineralized bone within the defect sites was determined by analysis of the micro-CT images by use of Tri/3D Bon software. As shown in Fig. 5b, the volume of newly formed bone in the CGF + BMSC group was greater than that of the CGF group, and that of the CGF group was greater than that of the control group. Significant differences between the groups were found 4, 8, and 12 weeks after operation except for between the CGF and CGF + BMSC groups at 12 weeks (n = 6 for each group/period).
Treatment of a rat calvaria critical-size bone defect with CGF and BMSCs: histological findings
Histological assessment of the osteogenic potential of CGF and CGF + BMSC was performed 4, 8, and 12 weeks after operation in the above-described rat calvaria defect model. Hematoxylin and eosin-stained (H&E) sections of bone were examined under a light microscope (Fig. 6a). In addition, we also analyzed 4-week samples by use of polarized light microscopy (Fig. 6b), because calcified immature bone tissue cannot be distinguished from collagenous fibrous tissue on decalcified H&E sections until several weeks after bone formation. Non-calcified woven bone is observed as shiny white fibrous tissue under a polarized light microscope [20, 21].

Treatment of rat calvaria critical-size bone defects with CGF and BMSCs: histological findings. a Representative light microscopic images of cranial tissue sections of the three groups, stained with H&E 4, 8, and 12 weeks after operation. (magnification ×20). Bar 1,000 μm. b Representative light (left column) and polarized light (right column) microscopic images of H&E stained cranial tissue sections of the three groups 4 weeks after operation, focused on the edges of the cranial holes (magnification ×40). Bar 1,000 μm. The yellow arrows indicate the edges of the cranial holes. The white arrows indicate woven bone tissue (shiny white fibrous tissue)
Histological analysis showed clear differences in tissue formation between the different treatment conditions. In the control group, sparse fibrous connective tissue was observed to traverse the empty defects and newly formed bone was only found in the area adjacent to the edges of the defect site 4 weeks after operation. Woven bone was not clearly observed in polarized microscopic images of the defect site. This pattern of bone formation was also observed after 8 and 12 weeks, except that the new bone at the periphery of the bone hole became thicker over time. Twelve weeks after operation, a large bone defect still remained in the control group.
In the CGF group, similar to the control group, only fibrous connective tissue was observed to traverse the defects and newly formed bone was observed at the edges of the defect site only 4 weeks after operation. However, in contrast with the control group, thin woven bone-like tissue was observed within the defects by polarized light microscopic analysis (Fig. 6b, white arrows). Over time, the new bone at the periphery gradually grew upwards to the central area of the defect site. Twelve weeks after operation, newly formed bone covered a large area of the bone defect.
In the CGF + BMSC group, islands of newly formed bone were observed 4 weeks after operation, which corresponded well with the micro-CT data obtained at this time. In contrast with the other two groups, newly formed bone was found both in the periphery and at the center of the defect site at this time. In addition, abundant neovascularization was observed around the new bone. The new bone became thicker over time, and, 12 weeks after the operation the critical-size bone defect was almost closed and bone marrow was confirmed to be present within the new bone tissues, resulting in a structure similar to that of normal cranial tissues.
Discussion
This study demonstrated that CGF, a second-generation platelet concentrate, effectively promoted the repair of bone defects and had particularly strong effects when combined with BMSCs. In a rat calvarial defect model, combined use of CGF and BMSCs almost completely repaired critical-size bone defects within 12 weeks. In addition, the CGF extract promoted the proliferation, osteogenic maturation, and mineralization of mesenchymal stem cells in vitro.
It is well established that in the rat calvarial defect model there is a gradual progression of new bone from the periphery of the bone hole to the center. New bone progresses in this direction because the cells that induce bone formation are mainly supplied from the cut edge of the bone defect. However, in our study, micro-CT and histological examination revealed new bone formation not only at the periphery of the bone hole but also in the central area of the bone defect in the CGF + BMSCs group, starting 4 weeks after the operation. This new bone included island-shaped new bone that had no contact with the cut edge of the periphery of the bone. Twelve weeks after the operation, critical-size bone defects were almost closed over in the CGF + BMSCs group, whereas large bone defects remained in the control group. This finding suggested that the fibrin network of CGF served as a scaffold for BMSCs that supported the formation of new bone by BMSCs, as reported by Dhohan, Choukroun et al. [2, 22, 23]. In addition, new bone mass significantly increased in the CGF and CGF + BMSCs groups starting 4 weeks after operation, compared with the control group, and this tendency continued until 12 weeks after operation.
Although very favorable results were obtained in these in-vivo experiments, the in-vitro effects of the CGF extract were different depending on the concentration of CGF used. The in-vitro experiments suggested that a highly concentrated CGF extract may inhibit osteogenic maturation and mineralization. Thus, whereas CGF extracts at concentrations between 1 and 10 % promoted cellular proliferation, osteogenic maturation, and mineralization in a dose-dependent manner, the effects of a 20 % CGF extract were inferior to those of the 10 % group, suggesting the existence of optimum doses of CGF. With regard to its effect on COL1A1 gene expression, expression of this gene decreased in the presence of a high concentration (20 %) of CGF compared with its expression in the control group (0 %), indicating that CGF might actually function to inhibit bone formation. We speculate that this inhibitory effect of CGF might be because, as reported by Dhohan et al. [4], the CGF extract contains not only growth factors favorable for bone formation but also inflammatory cytokines, for example tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1), which may inhibit bone formation (Fig. 4). Therefore, at a high concentration of CGF the inhibitory action of inflammatory cytokines on bone formation may dominate over the promoting action of the growth factors, which would result in unfavorable effects on bone formation. This concentration-dependent effect seems to be one of the reasons why contradictory effects of PRP on bone formation have been described in previous reports [8–10]. In addition, the results from polymerase chain reaction (PCR) analysis in this study did not reveal any significant change in the expression of such osteogenic master genes as RUNX2 and OSX, which determine whether MSCs are directed toward differentiation into osteoblasts, irrespective of the concentration of CGF extract. These results suggest that the CGF extract stimulates osteogenic maturation only and does not promote commitment of MSCs to osteoblast differentiation.
In this study, we found that CGF served as a good scaffold for BMSCs for treatment of a critical-size bone defect model. The advantages of using CGF as a scaffold are:
-
1.
CGF contains cytokines that promote cell growth, maturation, and matrix production;
-
2.
CGF preparation and cell integration into CGF, are quick and easy; and
-
3.
CGF is safe because it contains nothing but autologous blood ingredients.
For all these reasons we assume CGF will also work as a good scaffold for bone regeneration when it is applied clinically. However, when considering the actual clinical application of combination therapy of CGF and cell transplantation, it will be necessary to optimize the cells to be used (their origin and preparation) and the amount of CGF and other ingredients or modifications that must be applied.
Conclusion
CGF promoted the proliferation, osteogenic maturation, and mineralization of mesenchymal stem cells in vitro, and combination therapy of CGF with BMSCs enabled excellent healing of a critical-size bone defect in vivo. CGF in combination with mesenchymal cell transplantation may be suitable for treatment of large bone defects that are difficult to treat by other methods. However, further research and optimization of the cells and CGF used, and other conditions, are needed before clinical application.
References
Mosesson MW, Siebenlist KR, Meh DA. The structure and biological features of fibrinogen and fibrin. Ann N Y Acad Sci. 2001;936:11–30.
Dohan DM, Choukroun J, Diss A, Dohan SL, Dohan AJ, Mouhyi J, et al. Platelet-rich fibrin (PRF): a second-generation platelet concentrate. Part I: technological concepts and evolution. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:e37–44.
Dohan DM, Choukroun J, Diss A, Dohan SL, Dohan AJ, Mouhyi J, et al. Platelet-rich fibrin (PRF): a second-generation platelet concentrate. Part II: platelet-related biologic features. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:e45–50.
Dohan DM, Choukroun J, Diss A, Dohan SL, Dohan AJ, Mouhyi J, et al. Platelet-rich fibrin (PRF): a second-generation platelet concentrate. Part III: leucocyte activation: a new feature for platelet concentrates? Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:e51–5.
Brown LF, Lanir N, McDonagh J, Tognazzi K, Dvorak AM, Dvorak HF. Fibroblast migration in fibrin gel matrices. Am J Pathol. 1993;142:273–83.
Marx RE, Carlson ER, Eichstaedt RM, Schimmele SR, Strauss JE, Georgeff KR. Platelet-rich plasma: growth factor enhancement for bone grafts. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:638–46.
Pietrzak WS, Eppley BL. Platelet rich plasma: biology and new technology. J Craniofacial Surg. 2005;16:1043–54.
Plachokova AS, Nikolidakis D, Mulder J, Jansen JA, Creugers NH. Effect of platelet-rich plasma on bone regeneration in dentistry: a systematic review. Clin Oral Implant Res. 2008;19:539–45.
Freymiller EG, Aghaloo TL. Platelet-rich plasma: ready or not? J Oral Maxillofac Surg Off J Am Assoc Oral Maxillofac Surg. 2004;62:484–8.
Schmitz JP, Hollinger JO. The biology of platelet-rich plasma. J Oral Maxillofac Surg Off J Am Assoc Oral Maxillofac Surg. 2001;59:1119–21.
Dohan Ehrenfest DM, Rasmusson L, Albrektsson T. Classification of platelet concentrates: from pure platelet-rich plasma (P-PRP) to leucocyte- and platelet-rich fibrin (L-PRF). Trends Biotechnol. 2009;27:158–67.
Sohn DS, Heo JU, Kwak DH, Kim DE, Kim JM, Moon JW, et al. Bone regeneration in the maxillary sinus using an autologous fibrin-rich block with concentrated growth factors alone. Implant Dent. 2011;20:389–95.
Dohan Ehrenfest DM, de Peppo GM, Doglioli P, Sammartino G. Slow release of growth factors and thrombospondin-1 in Choukroun’s platelet-rich fibrin (PRF): a gold standard to achieve for all surgical platelet concentrates technologies. Growth Factors (Chur, Switzerland). 2009;27:63–9.
He L, Lin Y, Hu X, Zhang Y, Wu H. A comparative study of platelet-rich fibrin (PRF) and platelet-rich plasma (PRP) on the effect of proliferation and differentiation of rat osteoblasts in vitro. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;108:707–13.
Okamoto T, Aoyama T, Nakayama T, Nakamata T, Hosaka T, Nishijo K, et al. Clonal heterogeneity in differentiation potential of immortalized human mesenchymal stem cells. Biochem Biophys Res Commun. 2002;295:354–61.
Lucarelli E, Beccheroni A, Donati D, Sangiorgi L, Cenacchi A, Del Vento AM, et al. Platelet-derived growth factors enhance proliferation of human stromal stem cells. Biomaterials. 2003;24:3095–100.
Graziani F, Ivanovski S, Cei S, Ducci F, Tonetti M, Gabriele M. The in vitro effect of different PRP concentrations on osteoblasts and fibroblasts. Clin Oral Implant Res. 2006;17:212–9.
Weibrich G, Hansen T, Kleis W, Buch R, Hitzler WE. Effect of platelet concentration in platelet-rich plasma on peri-implant bone regeneration. Bone. 2004;34:665–71.
Choi BH, Zhu SJ, Kim BY, Huh JY, Lee SH, Jung JH. Effect of platelet-rich plasma (PRP) concentration on the viability and proliferation of alveolar bone cells: an in vitro study. Int J Oral Maxillofac Surg. 2005;34:420–4.
Sverzut CE, Lucas MA, Sverzut AT, Trivellato AE, Beloti MM, Rosa AL, et al. Bone repair in mandibular body osteotomy after using 2.0 miniplate system—histological and histometric analysis in dogs. Int J Exp Pathol. 2008;89:91–7.
Retamoso LB, Montagner F, Camargo ES, Vitral RW, Tanaka OM. Polarized light microscopic analysis of bone formation after inhibition of cyclooxygenase 1 and 2. Anat Record 2010;293:195–9.
Choukroun J, Diss A, Simonpieri A, Girard MO, Schoeffler C, Dohan SL, et al. Platelet-rich fibrin (PRF): a second-generation platelet concentrate. Part IV: clinical effects on tissue healing. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:e56–60.
Choukroun J, Diss A, Simonpieri A, Girard MO, Schoeffler C, Dohan SL, et al. Platelet-rich fibrin (PRF): a second-generation platelet concentrate. Part V: histologic evaluations of PRF effects on bone allograft maturation in sinus lift. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:299–303.
Acknowledgments
We thank Dr Junya Toguchida, Kyoto University, for kindly providing the hTERT-E6/E7 cells and Mrs Mari Shinkawa for technical assistance with the histological study. This study was supported by a Grant-in-Aid for Scientific Research (no. 23390364) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and by a special research subsidy from Terumo Life Science Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
No competing financial interests exist.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Honda, H., Tamai, N., Naka, N. et al. Bone tissue engineering with bone marrow-derived stromal cells integrated with concentrated growth factor in Rattus norvegicus calvaria defect model. J Artif Organs 16, 305–315 (2013). https://doi.org/10.1007/s10047-013-0711-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10047-013-0711-7