
Abstract
The geochemical processes, water–rock interactions and stable isotopes distribution (δ13C of DIC and δ18O and δ34S of \({\text{SO}}^{{{\text{2 - }}}}_{{\text{4}}} \)) were investigated in the gasoline-contaminated aquifer at the Hnevice site, 50 km northwest of Prague, Czech Republic. Diesel, gasoline and oil leaks originate from a large fuel storage area causing heavy contamination of the saturated and unsaturated zones in an area of about 0.7 km2. Groundwater investigations were conducted using five multilevel sampler wells with emphasis on redox parameters and degradation by-products and a solid-phase study focused on iron speciation and determination of principal and secondary minerals. Based on the study of groundwater and solid-phase geochemistry, four different geochemical zones were described. Zone I is thought to be background consisting of an aerobic aquifer and the absence of reduced species in significant concentrations. Zone II is situated in the plume core with methanogenic, sulphate and iron-reducing conditions accompanied by ankerite and kutnahorite precipitates and significant depletion of the oxidation capacity of the aquifer. Zone III is a mixing (corona) zone, situated at the fringe of the plume with high biodegradation rates and Fe(III)-precipitants. In zone IV, reoxidation of Fe(II) minerals (with e.g. the occurrence of psilomelane and cornelite) is typical.
Résumé
Les processus géochimiques, les interactions eau-roche et la distribution des isotopes stables (δ13C du DIC - Carbone Inorganique Dissout, du δ18O et du δ34S du \({\text{SO}}^{{{\text{2 - }}}}_{{\text{4}}} \)) ont été étudiés dans un aquifère contaminé par de l’essence, au site de Hnevice à 50 km au nord-ouest de Prague, en République Tchèque. Les infiltrations de diesel, essence et huile proviennent d’une grande aire de stockage de fuel, et sont la cause d’une contamination sévère des zones saturée et insaturé sur une surface d’environ 0.7 km2. Les études sur les eaux souterraines ont été réalisées en utilisant cinq puits d’échantillonnage multi-niveaux, et en portant une attention particulière aux paramètres redox et aux produits de la dégradation ainsi qu’à l’étude de la phase solide basée sur la spéciation du Fer et la détermination des minéraux principaux et secondaires. En se basant sur l’étude de la géochimie de l’eau souterraine et de la phase solide, quatre zones géochimiques différentes ont été décrites. La Zone I est supposée représenter un bruit de fond ; elle est caractérisée par un aquifère aérobic et par l’absence d’espèces réduites en concentrations significatives. La Zone II est située au cœur du nuage avec des conditions de méthanogénèse et de réduction des sulfates et du fer, accompagnées par des précipitations d’ankérite et de kutnahorite, et un appauvrissement considérable de la capacité d’oxydation de l’aquifère. La Zone III est une zone (en couronne) de mélange située sur la bordure du nuage, présentant de forts taux de biodégradation et des précipitations de Fe(III). Dans la zone IV la réoxidation des minéraux du Fe(II) est typique (avec la présence de psilomélane et de cornélite).
Resumen
Se han investigado los procesos geoquímicos, la interacción agua-roca y la distribución de isótopos estables (δ13C de CID, y δ18O y δ34S de \( {\text{SO}}_{{\text{4}}} ^{{{\text{2 - }}}} \)) en un acuífero contaminado por gasolina situado en Hnevice, a 50 km al noroeste de Praga, República Checa. A partir de un gran área de almacenamiento de combustibles se originaron vertidos de diesel, gasolina y aceites que causaron una fuerte contaminación de las zonas saturada y no saturada en un área de aproximadamente 0.7 km2. La investigaciones en las aguas subterráneas se llevaron a cabo usando cinco pozos de muestreo multinivel haciendo énfasis en los parámetros redox y los productos de degradación y el estudio de la fase sólida centrada en la especiación del Hierro y la determinación de los minerales principales y secundarios. Se describieron cuatro zonas diferenciadas geoquímicamente, basadas en el estudio geooquímico del agua subterránea y de la fase sólida. Se piensa que la Zona I representa el acuífero previo y consiste en un acuífero aerobio con ausencia de especies reducidas en concentraciones significativas. La Zona II está situada en el centro del penacho contaminante, con condiciones metanogénicas, sulfatos y hierro reducido acompañados por precipitados de ankerita y kutnahorita y un descenso significativo de la capacidad de oxidación del acuífero. La Zona III es una zona de mezcla (corona), situada en el borde del penacho con altos rangos de biodegradación y precipitados de Fe(III). En la zona IV es típica la reoxidación de minerales de Fe(II) (con la presencia de psilomelano y cornelita, por ejemplo).
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Many former army fuel storage areas exist in the Czech Republic. Handling petroleum products in these areas causes the release of water soluble and mobile products, like BTEX, to the environment and has created an important group of contaminated groundwater sites (Bennet et al. 1993; Chapelle 1999; Wiedemeier et al. 1999). Soil and shallow aquifers contain many species of microorganisms that can degrade a variety of organic compounds using a carbon source, electron donors and acceptors, as well as water and mineral nutrients to carry out their living functions. This process, leading to a decrease in contaminants and their complete removal from the aquifer, have been termed “natural attenuation” (Wiedemeier et al. 1999) and includes degradation, sorption, dilution, volatilization, precipitation and ion-exchange (Christensen et al. 2001). However, only microbial degradation really removes the mass of organic contaminants from the affected aquifer. Redox conditions are the main factors determining the potential and rate of degradation of organic contaminants; so therefore their characterization, understanding and evaluation at a site is essential for environmental risk assessment, for interpretation of the characteristics of groundwater samples from monitoring wells, and the determination of suitable remedial techniques. Monitored natural attenuation is beginning to be accepted as a useful alternative remediation technique to active remediation techniques.
Oxidized groundwater uncontaminated by the facility flows through the contaminated aquifer and brings electron acceptors (e.g. O2, \( {\text{SO}}_{{\text{4}}} ^{{2 - }} \), \( {\text{NO}}_{3} ^{ - } \)) to the contamination plume where they are used by bacteria to decompose hydrocarbons in the sequence: O2 (in water)-\( {\text{NO}}_{3} ^{ - } \) (in water)-Fe(III) (in solid phase)-Mn(IV) (in solid phase)-\( {\text{SO}}_{{\text{4}}} ^{{2 - }} \) (in water)-methanogenesis (Christensen et al. 2001). In the water flowing out of the contaminant plume the amounts of CO2, H2S, CH4, Fe(II), Mn(II), \( {\text{NH}}_{4} ^{ + } \), \( {\text{HCO}}_{{\text{3}}} ^{{\text{ - }}} \) and \( {\text{NO}}_{{\text{2}}} ^{{\text{ - }}} \) increase, electron acceptors are nearly depleted and consequently redox environments originate in the plume. Close to the source, zone methanogenesis occurs. Redox zonation continues sequentially downgradient with sulphate reducing and iron reducing zones as lower energy yield degradation processes which may be important in long-term pollutant attenuation and, when overlapping or simultaneously occurring is thermodynamically possible under a wide range of environmental conditions. This was observed in several field studies (Eganhouse et al. 1993; Postma and Jakobsen 1996; Heron et al. 1998; Cozzarelli et al. 1999; Van Breukelen et al. 2003). After that, zones of Mn (IV) and \( {\text{NO}}_{3} ^{ - } \) reduction occur, sometimes overlapping with the zone of iron reduction. At the border of the plume, aerobic conditions exist, with the concentrations of dissolved oxygen being higher than 1 mg/l (Christensen et al. 2000). This simplified vision of redox zones could be affected by several hydrogeological and geochemical factors (water-level fluctuations, changes in groundwater-flow direction, spatial heterogeneity in aquifer material) and in the field the redox zones do not occur individually and separately, but very often overlap.
The ability of the aquifer to restrict the development of reduced environments depends on the availability of oxidized species in the solid phase—Fe(III) minerals mainly—referred to as the oxidation capacity (OXC). If the oxidation capacity of the aquifer is low, large zones of very low redox levels will develop and methanogenic conditions will occur, while in the aquifer with high oxidation capacity, a sequence of different redox zones, dependent on the species contributing to the available oxidation capacity, will be present (Heron et al. 1994a). In the unsaturated zone, Fe and Mn are associated with Fe and Mn oxides and hydroxides in clay minerals and silicates. Iron oxides are reduced which leads to the increase of the Fe(II) ion in groundwater. This ion partly precipitates and partly migrates downgradient into more oxidized zones. In reduced environments, inorganic precipitates like FeS, FeS2 and iron and manganese carbonates originate; the oxidation capacity of the solid phase is reduced. In contrast, in aerobic conditions, Fe(II) oxidizes and precipitates as amorphous iron oxyhydroxides (Lovley 1997; Cozzarelli et al. 1999).
Laboratory studies have shown that petroleum hydrocarbons undergo isotopic fractionation during microbial degradation (Meckenstock et al. 1999; Ahad et al. 2000). As microbial degradation proceeds, the contaminant concentrations decrease and the residual fraction is being enriched by heavier isotopes, so the 13C/12C ratio in the residual substrate fraction is increased. In closed systems, the isotope fractionation process due to microbial degradation can be described by the carbon isotope fractionation factor (Cα) and the Rayleigh equation and the Cα can be used to calculate the amount of compound biodegraded when the initial concentration of the contaminant is known (Meckenstock et al. 1999; Richnow et al. 2003). If methanogenesis is the main biodegradation process in the reduced parts of the contaminated zone, methane generated during CO2 reduction is enriched in the lighter isotope (12C). The CO2 associated with this methane production is enriched in the heavier isotope (13C) (Hornibrook et al. 2000). The δ13C-DIC (dissolved inorganic carbon) in the plume is enriched with respect to background. High values are generally observed and are thought to be related to production of isotopically positive CO2 by methanogenesis (Hackley et al. 1996) and to a lesser extent by outgassing of CO2 (Hornibrook et al. 2000).
If microbial sulphate reduction contributes to the removal of petroleum hydrocarbons, this process is associated with significant isotopic effects for both S and O in sulphate. During the occurrence of dissimilar sulphate reduction processes, bacteria reduce sulphate to sulphide to obtain energy for the oxidation of carbon sources and sulphur is fractionated by a kinetic mechanism with preferential incorporation of 32S to a sulphide product. When biological sulphate reduction occurs, oxygen isotopic exchange between sulphate and groundwater is promoted. In most natural situations, sulphate is depleted in 18O relative to equilibrium with groundwater and causes an enrichment of 18O in the residual sulphate. If sulphate reduction proceeds, the resulting reaction product is lighter due to kinetic fractionation and the rest of the dissolved sulphate is isotopically heavier with more positive values of 18O and 34S (Chambers and Trudinger 1979). Recently, the isotopic fractionation of organic contaminants is being used as a tool for demonstrating in situ biodegradation of petroleum hydrocarbons in many laboratory experiments (Meckenstock et al. 1999; Ahad et al. 2000; Wilkes et al. 2000; Bolliger et al. 2001) and for obtaining quantitative information on microbial reduction at many contaminated sites (Strebel et al. 1990; Bottrell et al. 1995; Schroth et al. 2001; Mancini et al. 2002; Richnow et al. 2003; Van Breukelen et al. 2003; Meckenstock et al. 2004).
The concentrations of the petroleum hydrocarbons, electron acceptors and degradation by-products have been monitored during 2003–2004 in multilevel sampler wells at the Hnevice site. The purpose of this study was to characterize redox conditions in groundwater, describe changes in the distribution of stable isotopes and investigate the chemical behaviour of iron and changes in the mineralogical composition of the affected aquifer. In contrast to a large number of papers on the natural attenuation of contaminants, this study was carried out at the upstream end of the contaminant plume.
Site characteristics
Site description
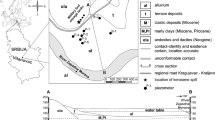
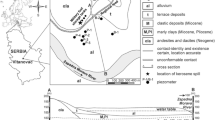
The Hněvice test site is situated 50 km northwest of Prague in the Czech Republic, near Roudnice nad Labem (Fig. 1a). A large fuel storage area located there has been used since 1943 and caused contamination in the unsaturated zone of the Quaternary aquifer and in the groundwater. Sources of contamination included leakage from corroded fuel-storage tanks and pipelines, and transport of petroleum hydrocarbons between rail sidings and storage tanks. Most of the stored products consist of gasoline, kerosine and diesel and a smaller volume of oil. The spectrum of contaminants is wide and the most studied were petroleum hydrocarbons and mainly the included BTEX (benzene, toluene, ethylbenzene and xylenes). The exact quantity of escaped products is not known, but according to the hydrogeological surveys, there should be thousands of tons of petroleum hydrocarbons in the subsurface rock environment. The area of interest covers about 2.5 km2 and is only about 350 m from the River Labe (Elbe). The current extent of the contaminant plume area is about 0.7 km2 and its mobility is very low. Stratification of groundwater pollution is significant: high concentrations of dissolved petroleum hydrocarbons are in the upper unsaturated layers and concentrations at the aquifer base are close to background. The fuel-free layer is in a strip parallel to the River Labe and has been shrinking since the maximum amounts of petroleum hydrocarbons were detected in the 1970s and 1980s. Total petroleum hydrocarbons (THP) in water varied from about 4 mg/l in the west to <0.3 mg/l in the east. The locations of sampled wells and contaminant plume borders are shown in Fig. 1b.

a Location of the Hnevice site in the Czech Republic and in Europe. b Location of the monitored wells
The geological basement of the Hnevice site is in the western part of the Bohemian Cretaceous Basin and consists of Proterozoic crystalline rocks, Permocarboniferous strata and Cretaceous strata represented by the Peruc–Korycany Formation and the Bílá Hora Formation. Proterozoic rocks are represented by amphibolitic diorites with spessartite veins or biotitic granite. The Peruc–Korycany Formation was deposited in brackish and saline water and consists mainly of sandstones. The Bílá Hora Formation consists of sandy and calcic claystones, siltstones, and silty sandstones and has the hydrologic character of a regional semi-aquitard. Quaternary unconsolidated strata, which contain petroleum hydrocarbon contaminants, are represented by the River Labe terraces with a maximum thickness of 14.5 m. They consist of fine sand with gravel layers and boulders (granular texture is coarser with depth) and, in upper parts, of clayey over sandy flood-deposited-soils. A geological map and cross sections of the studied area are illustrated in Fig. 2a–c (from Hösl 2003).


Previous hydrogeological investigations show the presence of two aquifers at the Hnevice site—the Quaternary fluvial aquifer (upper aquifer) and the Cenomanian (Cretaceous) aquifer (lower aquifer). The Quaternary aquifer is in the Labe River terrace, and the Cenomanian aquifer is in the Peruc–Korycany sandstones (Herèík et al. 1999). The direction of groundwater flow in both aquifers is to the E–NE, to the Labe River, which drains the whole area. The lower aquifer is confined with dual porosity and its hydraulic conductivity ranges from n×10−6 to n×10−5 m/s and its piezometric surface is higher than the groundwater level of the upper aquifer. This head relationship between the two aquifers protects the lower aquifer from contact with contaminated water from the upper aquifer. The upper aquifer has a high intergranular porosity with the hydraulic conductivity ranging from n×10−4 to n×10−3 m/s, a transmissivity value of about 1,800 m2/day and an effective porosity 0.25. The groundwater level is 4–6 m below ground level (bgl) and the maximum saturated thickness is about 10 m (Hazdrová et al. 1980).
Materials and methods
The geochemical heterogeneity and distribution of stable isotopes in groundwater and the iron speciation and mineralogical composition in the solid phase of the affected aquifer were studied in 91 samples from the aquifer. The geochemical data were collected from five permanent multilevel sampler wells (MLS); HJ506, HJ507, HJ508, PJ519, PJ520 during fieldwork conducted in 2003–2004 (Fig. 1b). For background; one older well and one MLS borehole, PJ500 and HJ506 respectively, were used to define the area of entrance of groundwater to the contaminated area. Downstream to the contaminant plume, wells HJ507 and HJ508 have been installed in order to monitor groundwater quality at the border of the contaminated area. Well PJ519 in intended to monitor the plume fringe and well PJ520 is intended to monitor the plume core. To examine the small-scale geochemical heterogeneity of the studied aquifer, sampling was done at discrete depths in the range 15–50 cm in groundwater and 25–50 cm in the solid phase; 44 water samples and 47 aquifer material samples were so obtained. The code for each multilevel sample consists of the sampler well number and sampling point number, e.g. PJ520/5. The number of the sampling ports starts at 5 m bgl.
Groundwater sampling and analysis
Analysis of groundwater was done with emphasis on redox parameters and degradation products (pH, Eh, TPH, O2, CO2, H2S, CH4, Fe, Mn, \( {\text{NH}}_{{\text{4}}} ^{{\text{ + }}} \), \( {\text{SO}}_{{\text{4}}} ^{{{\text{2 - }}}} \), \( {\text{HCO}}_{{\text{3}}} ^{{\text{ - }}} \), \({{\rm{NO}}_{{\rm{3}}} ^{{\rm{ - }}} }\), \({{\rm{NO}}_{{\rm{2}}} ^{{\rm{ - }}} }\), TOC).Water samples were collected with a footvalve pump.
Temperature, pH, Eh, electrical conductivity and dissolved oxygen were detected on site with a combined WTW pH/Eh-meter. Alkalinity and acidity were detected immediately after sampling by potentiometric titration. Amounts of \({\text{HCO}}^{{\text{ - }}}_{{\text{3}}} \)were calculated from alkalinity titration. Samples for Fe and Mn measurements were filtered through 0.45-μm-membrane filter, fixed by super pure 0.1 M HCl and stored in 250-ml-PE bottles. Samples for H2S analysis were sampled to 40-ml-dark vials and fixed by NaOH and CdCl2. Samples for \( {\text{NO}}_{{\text{3}}} ^{{\text{ - }}} \), \( {\text{NO}}_{{\text{2}}} ^{{\text{ - }}} \), CO2 forms, \( {\text{HCO}}_{{\text{3}}} ^{{\text{ - }}} \), Ca, Mg, Na, K, Cl− and F− were taken to 1l PE bottles.
For TPH analysis samples were taken into 1l dark glass bottles. Samples for TOC were taken into 250-ml-PE bottles. Water samples for BTEX, CH4, ethylbenzene, isopropylbenzene, n-propylbenzene, ethyltoluene, 1,3,5-trimethylbenzene, 1,2,4-trimethylbenzene and 1,2,3-trimethylbenzene were taken in 40-ml-glass vials and capped carefully.
All samples were stored at 4°C during transport to the laboratory. Analyses were carried out immediately after sampling on site (pH, Eh, temperature, electrical conductivity, alkalinity, acidity) or within 24 h (nitrogen and sulphur species, CO2 forms, \( {\text{HCO}}_{{\text{3}}} ^{{\text{ - }}} \)) or within 1 week (metals, TOC, TPH, BTEX, CH4, F−, Cl−).
The concentrations of Ca, Mg, Na, K, Fetotal and Mntotal were detected by atomic absorption spectrometry (AAS). Analyses for Fe(II) were done spectrophotometrically by reaction with 1.10-phenantroline. Species of nitrogen were detected spectrophotometrically, \( {\text{NH}}_{{\text{4}}} ^{{\text{ + }}} \) ions by salicylate method, \( {\text{NO}}_{{\text{3}}} ^{{\text{ - }}} \) by reaction with sulphosalicyle acid and \( {\text{NO}}_{{\text{2}}} ^{{\text{ - }}} \) by reaction with NED-dihydrochloride. Detection of Cl− ions was by argentometric titration (Mohr method). Sulphate was detected gravimetrically by reaction with BaCl2. Sulphides and hydrogensulphide were analysed spectrophotometrically. Detection of TPH was by FTIR (Fourier transform infrared) spectrophotometry, and concentrations of BTEX, CH4, ethylbenzene, isopropylbenzene, n-propylbenzene, ethyltoluene, 1,3,5-trimethylbenzene, 1,2,4-trimethylbenzene and 1,2,3-trimethylbenzene were measured by gas chromatography with flame ionization detector (GC-FID). TOC was measured by thermal oxidation-NDIR (non-dispersive infrared).
For δ13C-DIC measurements, samples were taken into 100-ml-glass bottles and I2-KI solution was added. Analysis was performed in laboratories of the Czech Geological Survey, Prague. For analysis of δ18O and δ34S in sulphate samples were made alkaline (pH 10) and filtered to remove iron. Then the samples were acidified to pH 2.0–2.5 and heated to approximately 80°C on a hot plate. A volume of 100 g/l BaCl2 solution equal to 10% of the sample volume was added and the BaSO4 precipitate was left covered overnight to cool and coarsen. The barium sulphate precipitate was recovered on a 0.45-μm-membrane filter and washed with deionized water prior to drying at 50°C. Analysis was finished in the Environmental Engineering Research Centre, School of Civil Engineering, Queen’s University Belfast (Northern Ireland), on DELTA PLUS TC/EA and Micromass Eurovector TC/EA.
Solid-phase sampling and analysis
Analysis of the solid phase of the aquifers was focused on iron speciation and determination of the principal minerals. Extractions of 6 M HCl hot and Ti-EDTA were carried out, replenished by carbonate content analysis and supplemented by microscopical observations, X-ray diffraction, and scanning electron microscopy (SEM).
Sediment samples were taken during drilling in the winter of 2003–2004. Because of presumptive microbial activity, short holding times were preferred in all chemical determinations and only the centres of the cores were used. Immediately after drilling, all samples were deposited in 40-ml-glass vials, pressurized with nitrogen to exclude oxygen from the air, sealed by wax and placed in the freezer.
Methods for detailed speciation of Fe and S minerals in aquifer solids were performed on triplicates of sediment samples from the Hnevice site, using methods developed by Heron et al. (1994a, b) and Heron and Christensen (1994). This set of chemical extractions provides a speciation of the total iron content and ferrous and ferric iron speciation. Oxidation capacity of the sediment was quantified using a reductive Ti(III)-EDTA extraction. For total amount of Fe, hot 6 M HCl extraction was performed for 1 h. Species of Fe(II) were consequently measured spectrophotometrically. The Fe(II) sulphide species were distinguished as acid volatile sulphide (AVS) with 6 M hot HCl extraction and sequential HI and Cr-HCl pyrite extraction (detection limit 1 mg/g). In all extracts, Fe and Mn amounts were detected by AAS. The carbonate content in Hnevice samples was measured after sinter decomposition determined by alkalimetric titration (performed by MEGALAB Ltd., Prague).
Powder X-ray diffraction (XRD) analyses and scanning electron microscopy investigations (SEM) of all samples were performed at the Institute of Geochemistry, Mineralogy and Mineral Resources, Faculty of Natural Sciences of Charles University in Prague. Analysis of XRD was performed with a X’Pert Pro system PANanalytical B.V. using a Cu anode. The routine operating conditions were 40 kV/30 mA, step scanning at 0.05/30 s in the range 3–70 2 “theta”. Peak identification on the diffractograms was performed using the X’Pert HighScore 1.0d (PANanalytical B.V., Almelo, The Netherlands) data analysis software package. Samples for SEM investigation were imbedded in a raisin roller and coated with carbon. SEM imaging and analyses were performed using the electron microscope Cam Scan S4 with energy-dispersive analyzer EDX LINK ISIS 300, calibrated with standard SPI#02753-AB with a downloading time of 60 s.
Results
In the upper part of the aquifer, the hydrocarbons plume resulting from the leaks has been detected in a zone 5–8 m bgl. In the spring of 2003, the composition of hydrocarbon species was studied in HJ508 and the following main constituents were found in the groundwater samples: ethylbenzene (0.09 mg/l), xylenes (0.47 mg/l), isopropylbenzene (0.029 mg/l), n-propylbenzene (0.042 mg/l), ethyltoluene (0.33 mg/l), 1,3,5-trimethylbenzene (0.076 mg/l), 1,2,4-trimethylbenzene (0.34 mg/l), 1,2,3-trimethylbenzene (0.17 mg/l), all confirmed at depths of 5.5–6.5 m bgl. The petroleum hydrocarbons contamination was found in sediment samples in well HJ508 (0–6.44 mg/kg of dry weight sediment), PJ519 (21–6,600 mg/kg) and PJ520 (45–8,800 mg/kg). The presence of isopropylbenzene (0.14 mg/kg), n-propylbenzene (0.31 mg/kg), ethyltoluene (1.83 mg/kg), 1,3,5-trimethylbenzene (0.58 mg/kg), 1,2,4-trimethylbenzene (1.7 mg/kg), 1,2,3-trimethylbenzene (0.25 mg/kg), n-heptane (0.66 mg/kg), cyclohexane (0.2 mg/kg) and methylcyclohexane (0.77 mg/kg) was detected in solid phase samples of HJ508. TPH contamination in sediment samples fit into a narrow zone 4–6.5 m bgl.
Hydrochemistry
Uncontaminated background groundwater at Hnevice site (represented by wells PJ500 and HJ506) has nearly neutral pH, stable \( {\text{HCO}}_{{\text{3}}} ^{{\text{ - }}} \) content (approx. 317 mg/l) and equilibrium with calcite. Groundwater uncontaminated by the test site is rich in nitrate (133 mg/l) and sulphate (310 mg/l) due to agricultural activities and the presence of a power station ash landfill near the test site area, and has measurable amounts of dissolved oxygen, a low amount of iron and concentrations of methane and sulphides below the detection limit (Table 1).
Downstream to the contaminant plume, wells HJ507 and HJ508 contain small amounts of petroleum hydrocarbons (up to 1.4 mg/l in the upper part of the aquifer). During the sampling event in the winter of 2004, water analyses show a significant decrease in nitrate concentrations and small decreases in sulphate. The concentrations of DIC, Fe and Mn are stable and amounts of reduced species like methane and sulphides are below the detection limit. Total petroleum hydrocarbons concentration in PJ519 was approx. 5 mg/l. Nitrate concentration decreased with respect to background to 77 mg/l and nitrite concentration increased to 4.72 mg/l. The same trend shows sulphate with decreases to 190–290 mg/l. Increase with respect to background was detected in iron (5.47 mg/l) and manganese (1.15 mg/l) content. Methane and sulphides concentrations were below the detection limit.
The most contaminated part of the Hnevice test site represented by PJ520 contains xylenes, ethyltoluenes and trimethylbenzenes mainly with maximum total petroleum hydrocarbons in the amount of 4 mg/l. TPH contamination reaches a depth of about 7 m bgl and has some traces of nitrate and sulphate, but much lower than background. Nitrate was detected in concentrations of about 15–32 mg/l with a steady increase of nitrite to 3.54 mg/l amounts of sulphate decreased to 80 mg/l with synchronical increase of sulphides to 6.2 mg/l. Amounts of reduced species like Fe(II), Mn(II) and CH4 increased to measurable concentrations and in the upper part of PJ520 (5–7 m bgl) and a significant increase of \( {\text{HCO}}_{{\text{3}}} ^{{\text{ - }}} \) was observed (Table 1). Vertical distribution of electron donors and acceptors in multilevel samplers at the Hnevice site are shown in Fig. 3a–d.

Vertical distribution of electron donors and acceptors in multilevel samplers PJ519 and PJ520 at Hnevice site: a oxidized species; b Fe and Mn; c reduced species; d alkalinity
Solid phase geochemistry
Aquifer material uncontaminated by the test site (wells HJ506, HJ507) consists of fluvial sand to gravelly sand, is low in organic matter, iron oxides (total iron amount <1% by weight), sulphides (<1 mg/g FeS) and carbonate content (0–0.07% dry mass), with the oxidation capacity value about 30–60 μeq/g (microequivalent/gram). Primary iron minerals are present as solid grains of crystalline iron oxides (magnetite, hematite, goethite) or amorphous ochres (e.g. limonite). Some amounts of iron are present in silicates, e.g. micas and amphiboles. Manganese is present in oxides like manganite and pyrolusite.
TPH contamination occurs in soil in wells HJ508 (below detection limit (BDL)−6.44 mg/kg), PJ519 (44–6,600 mg/kg), and PJ520 (11–8,800 mg/kg), with significant amounts of TPH in the horizon from 4.5–6.5 m bgl in HJ508 (Table 2). In the saturated zone in HJ508, the oxidation capacity (OXC) decreases with respect to background to values 17–30 μeq/g and the soil becomes enriched in iron to a maximum of 7,100 mg/kg of total Fe and 1,300 mg/kg of Fe(II). Amounts of sulphides and carbonates in the solid phase are below the detection limit. Mineralogical investigation lead to confirmation of iron and manganese oxides, hydroxides and sulphate (e.g. goethite, magnetite, hematite, limonite, psilomelane and cornelite) present like individual grains or cementating quartz and feldspar grains. Enrichment in iron content continues downstream to the plume in the upper parts of PJ519 with values of 2,300–7,700 mg/kg of Fetot and 1,410 mg/kg of Fe(II) (6 M HCl extraction values, Table 2). The oxidation capacity is lower with respect to background or HJ508 (17–25 μeq/g). In PJ519, no carbonate or sulphide precipitates were detected. Secondary iron and manganese oxides and hydroxides were confirmed by X-ray diffraction and SEM (e.g. goethite, magnetite, hematite, Table 2, Fig. 4d).

SEM photographs of iron and manganese containing species from the Hnevice site plume. a Ankerite in reactive belt of a feldspar grain (zone II); b zonal kutnahorite grain (zone II); c XRD spectrum of kutnahorite grain shown in b; d iron oxyhydroxides cementating the quartz grains (zone IV)
In the upper part (4.5–5.5 m bgl) of PJ520, iron and manganese carbonate precipitates (ankerite and kutnahorite) were confirmed both by X-ray diffraction and SEM (Table 2; Fig. 4a–c). Precipitates are present in the form of coatings of quartz grains or like individual grains with size 10–50 μm. Carbonate precipitation is evident also via increase of \( {\text{CO}}_{{\text{3}}} ^{{{\text{2 - }}}} \) content to 0.022% of dry mass. In the horizon 6–7.75 m bgl, the presence of secondary iron oxides (e.g. hematite, goethite and magnetite) is typical. The total iron amount from 6 M HCl extraction varies in range from 2,050–6,100 mg/kg. Fe(II) participates in this amount with a content of 350–950 mg/kg and both maximums occur at the depth of 4.5 m bgl. The oxidation capacity (OXC) of the sediment in PJ520 is low (7–15 μeq/g) in the upper part of the well (4–5.5 m bgl) and increases to values about 20–37.6 μeq/g at 6–7.75 m bgl. The amount of sulphides in all samples is below the detection limit and no sulphide presence was detected by X-ray diffraction or SEM.
Isotope geochemistry
Eight samples were investigated for δ13C in DIC and seven for δ18O and δ34S in sulphate in wells PJ500 and HV43 as background, along with samples from HJ508/5, HJ508/7, PJ519/5, PJ519/14 (δ13C-DIC only), PJ 520/6 and PJ520/16 from the contaminated area.
In groundwater at this site, a range of δ13C-DIC was found between −7.66 and −15.58‰ PDB and the \( {\text{HCO}}_{{\text{3}}} ^{{\text{ - }}} \) ion concentration varied between 317 mg/l in background to 634 mg/l in the upper part of PJ520 (Table 3). Samples from contaminated area show values δ13C-DIC from −13.44‰ PDB to −15.11‰ PDB, with exception of sample PJ520/6 (δ13C-DIC value −7.66‰ PDB).
Sulphate concentrations were decreasing in a direction into the contamination plume from 310 mg/l in PJ500 to 270 mg/l in HJ508 and PJ519 and 170 mg/l in the upper part of PJ520 (Table 3). Samples show δ34S values varying from 31 to 36‰. In the background, δ34S values are about 34–35.8‰. With decreasing concentrations of sulphate in the internal part of the plume, δ34S increases to a value of 36‰ with the exception of sample PJ520/6 (value 31‰). The values of δ18O in sulphate are between 10.1–11.74‰.
Discussion
Groundwater geochemistry
In the period 2002–2004, the plume was a subject of detailed geochemical studies. A total of 96 groundwater samples were collected for determination of major inorganic and organic species and 47 solid samples were investigated for primary and secondary iron and manganese mineral species and total petroleum hydrocarbons content. The data on concentrations of the electron acceptors and degradation by-products in the aquifer were used to study evidence of natural attenuation processes. Five MLS were installed upstream of the plume, in the fringe, where ambient groundwater containing high concentrations of electron acceptors enter the contaminated area and in the plume core.
The concentration of TPH increased in the direction towards the source zone reaching sampling points HJ507, HJ508, PJ519 and PJ520. Downstream to the contaminant plume, sampling points HJ507 and HJ508 contained small amounts of petroleum hydrocarbons (up to 1.4 mg/l in the upper part of the aquifer). The most contaminated part of the Hnevice test site represented by PJ520 contained xylenes, ethyltoluenes and trimethylbenzenes mainly with a maximum total petroleum hydrocarbons concentration of 14 mg/l. TPH contamination reaches to 7 m below ground level.
The zone of high concentrations of dissolved hydrocarbons coincided with maximum concentrations of the hydrocarbons determined in the sediment samples. The depletion zones of the electron acceptors and increase of amounts of degradation by-products correlate well with changes in hydrocarbon contamination. A full sequence of redox zones that sometimes overlap at many sites polluted with organic compounds (Heron et al. 1998; Cozzarelli et al. 1999; Christensen et al. 2001) has been identified at Hnevice site with degradation processes in vertically thin zones (up to 1 mm). Nitrate reduction causes decrease of nitrate downstream to the plume—nitrate reduction is in process at the mixing zone of the contaminant plume and nitrate containing groundwater in upper parts of HJ507, HJ 508 and PJ519 (5–6 m bgl). Manganese reduction at Hnevice site prevailed in the upper part of PJ519, where concentration ratio Fe/Mn is 1/2. In solid phase and background groundwater, Mn content was 1 order of magnitude lower than Fe(II) content. Evidence of iron reduction in the plume was found in PJ519 and partly PJ520. Elevated Fe(II) concentrations increasing in the direction to the plume correlate with a higher amount of secondary iron (hydr)oxides and carbonates content in the plume. The sulphate reduction zone coincides with maximum concentration of the dissolved hydrocarbons in PJ520 accompanied by occurrence of sulphides in a narrow zone close to the water table. In the same depth of PJ 520, methanogenesis occurs with high methane concentrations between 5–6 m bgl. Spatial distribution of the electron acceptors and degradation by-products in the PJ519 and PJ520 is illustrated in Fig. 3a–d.
Solid phase
Significant evidence of iron reduction in the plume was found. The oxidation capacity (OXC) of aquifer sediment, which is related to iron and manganese oxides and hydroxides, is the lowest near to the plume core and the highest values were found in the background. This indicates a depletion zone due to iron reduction close to the plume core zone followed by zones where iron has been precipitated and oxidized. The location of the zone in which Fe(III) is lowered corresponds well with the 6 M HCl extraction showing an increase in Fe(II). By studying groundwater geochemistry, this zone is thought to be methanogenic and sulphate reducing, overlapped from the inflow site of the plume by a Fe(III) reducing zone.
The dissimilar bacterial reduction processes influence the aqueous geochemistry and mineralogy of the contaminant plume region and the amount of dissolved Fe(II) increases in the region of Fe(III) reduction. In parallel, the concentration of extractable sedimentary Fe(III) oxides decreases. Extractions of 6 M HCl show that Fe(III) concentrations are higher than Fe(II) in all samples from the test site. Most abundant enrichment is obvious close to the most reduced zone (zone II) where Fe(II) content is about 50% of the total amount of iron.
Geochemical equilibrium calculations shows that siderite and ferroan calcite precipitation may occur in the anoxic zone (Table 4) and SEM confirmed the presence of iron and manganese carbonates as described in some previous studies (Tucillo et al. 1999; Van Breukelen et al. 2003). Iron and manganese carbonates (ankerite/kutnahorite) are present as distinct crystals or coatings on quartz and feldspar grains in the plume core. These minerals are possibly precipitates from the development of the reduced plume, as indicated by geochemical equilibrium calculations showing supersaturation with respect to siderite. Distribution of these mixed carbonates is related to the layer where low oxidation capacity connected to Fe(III) oxides was observed. Reduction of the Fe(III) is indicated in the layer accompanied with Fe(II) carbonates precipitation. Carbonate precipitation, which buffers the Fe(II) concentration released by iron reduction, was also confirmed by silicate analysis with increase of carbonates concentration from below the detection limit (BDL) to 0.02% of dry mass (Table 2).
The distribution of reduced sulphur species was determined by 6 M HCl sequential extraction. Although the presence of FeS or FeS2 is probable due to geochemical equilibrium calculations in all samples from the Hnevice test site, the content of sulphides was below the detection limit. The presence of sulphur in solid phase was confirmed by SEM investigations on the border of the core zone and fringe of the plume. There is no sulphur in individual form or pyrite/mackinawite crystals as described in many case studies (e.g. Cozzarelli et al. 1999; Tucillo et al. 1999), but iron oxyhydroxide grains contain small amounts of sulphur. This could be caused by reoxidation of the produced sulphides in the plume. Also the accumulation of H2S in the presence of high amounts of Fe(II) indicates that equilibrium processes may not control geochemical conditions in the anoxic zones. There may be some kinetic inhibition that limits high amounts of this precipitate or the influence of relatively high advective flow (1 m/day) moves precipitation reactions deeper in to the plume.
Isotope geochemistry
Typical geochemical zonation of the contaminant plume can be confirmed by carbon isotope fractionation investigations. The background groundwater is in equilibrium with calcite (average values about −12.7‰ PDB), the reoxidation zone and plume fringe are typified by redox reactions with petroleum hydrocarbons and the plume core is significantly enriched in δ13C-DIC by methanogenesis. Samples from the reoxidation zone and plume fringe show a tendency toward enrichment in δ13C-DIC in the residual fraction in the plume fringe (Van Breukelen et al. 2003) with values from −13.44‰ PDB to −15.11‰ PDB. Biodegradation creates additional DIC (CO2) and shifts original values of δ13C-DIC to more negative values and the value −7.66‰ PDB from PJ520/6 shows presence of methanogenesis. During methanogenesis, 12C is incorporated into methane and DIC evolved in the reaction is being enriched at the site in δ13C up to +38‰ PDB.
Groundwater sampling in multilevel wells has identified the same geochemical zones as were confirmed by investigation of δ18O and δ34S of sulphate. According to the literature (Spence et al. 2001), lower sulphate concentrations should be associated with high δ34S values and should show a linear trend characteristic for bacterial sulphate reduction. This inverse correlation trend is evident in the plume fringe and reoxidation zone. Values of δ34S are about 35.8‰ and with decreasing concentrations of sulphate in reoxidation zone δ34S increases to a value of 36‰.
Sample PJ520/6 from the plume core zone (δ34S value 31‰) is not enriched in heavier isotopes and also 18O does not show any strong enrichment in residual sulphate during bacterial reduction with values between 10.1 and 11.74‰. Processes could probably be affected by two factors—sorption and changes in advective flow velocity. In the soil environment a large amount of iron and manganese oxyhydroxides is being precipitated causing a large sorption capacity. It could bind sulphate ions and with a change of pH, release them causing an erroneous interpretation. At the depth of 5.5 m bgl, where sample PJ520/6 was taken, there is also a layer with higher advective flow that could affect the isotope distribution process by dilution.
Conceptual model of the test site
Due to 30-years of natural attenuation processes, the absence of any new spills and advective flow of the groundwater, the forehead of the contaminant plume at Hnevice site has moved to the north. The plume is shrinking from its maximum in the 1970s and 1980s (see Fig. 1), and geochemical conditions in the affected aquifer have changed. Based on groundwater and solid-phase geochemistry data obtained during fieldwork in 2003–2004, a conceptual model with four different geochemical zones was developed (Fig. 5). Zone I is thought to be the background with an unpolluted aerobic sandy aquifer (hematite, goethite, primary magnetite and pyrolusite were confirmed by SEM to be present in the solid phase), neutral pH, oxidized water rich in electron acceptors and no significant presence of reduced species from natural attenuation processes. Aerobic conditions are present in zone I followed by a zone of nitrate reduction. High amounts of nitrate are present in the groundwater at the Hnevice site (>100 mg/l in the background) due to agriculture facilities near the test area, so even in sulphate and iron reducing zones the nitrate is not completely depleted.

Conceptual model of the test site, based on fieldwork 2003–2004. The vertical lines represent the monitoring wells
Zone II, situated in the plume core, is the most contaminated part of the plume with a thick free phase layer of contaminants (i.e. connected body of non-aqueous-phase liquids situated on the water table that migrates through the subsurface on the water table). Electron acceptors like oxygen, nitrate and Mn(IV) are nearly consumed here and methanogenesis with sulphate and Fe(III) reduction zones occur. Saturation index calculations in PHREEQC (Table 4) showed that rhodochrosite and siderite will precipitate and the water is supersaturated also with respect to calcite, but in fact mixed carbonates like ankerite FeCa(CO3)2 and/or kutnahorite MnCa(CO3)2 precipitate. Coating of iron oxide surfaces with reduced iron phases in zone II appears to limit iron reduction and makes sulphate reduction and methanogenesis occur in the presence of biologically-available Fe(III). In zone II and partly in zone III, denitrification, manganese, iron and sulphate reduction occur where zones of iron and sulphate reduction overlap. The biogeochemical reactions and resulting chemical composition of shallow groundwater at the Hnevice site are affected by both microbially mediated redox reactions and heterogeneities in the composition and permeability of the aquifer material. Degradation of petroleum hydrocarbons occurs on a small scale in this part of the aquifer and creates small-scale geochemical changes in a 50-cm layer of relatively low permeability. In this microenvironment, conditions are favourable for sulphate reduction and methanogenesis in the presence of some amount of nitrate and biologically-available Fe(III).
Zone III is a mixing (corona) zone situated at the fringe of the plume. It is characterized by typically high biodegradation rates and the precipitation of secondary phases of Fe(III). Also nitrate, manganese and partly iron reducing zones overlap here. In the fringe of the plume, Fe(II) ions from reducing zone migrate to the more oxidized parts of the plume and iron oxides and hydroxides (e.g. ferrihydrite, goethite) precipitate.
Zone IV is a reoxidation zone between the core zone, the fringe of the plume and the background. Its existence is connected with movement of the forehead of the plume, where anaerobic conditions change back to aerobic conditions and/or nitrate reduction. In the solid phase (as confirmed by X-ray diffraction and SEM investigations), Fe(II) minerals are reoxidizing and species like ferrihydrite, manganite, cornelite (Fe2(SO4)3×7.5H2O) and psilomelane are generated. This reoxidation at the forehead of the plume makes a barrier for electron acceptors entering the plume because some amounts of them are consumed before they can participate in the biodegradation processes.
Conclusions
Groundwater geochemistry, stable isotopes distribution and sediment geochemical properties were studied at the inflow part of a contaminant plume at the Hnevice site in a Quaternary aquifer consisting of sand with gravel layers and boulders. Degradation of petroleum hydrocarbons occurs in discrete zones (cm scale) and a full sequence of redox zones (sometimes overlapping) as described at other sites (Heron and Christensen 1995; Cozzarelli et al. 2001), has been identified in this plume. Four zones in a hypothetical conceptual model of the Hnevice site have been confirmed:
-
I.
Background groundwater with high concentrations of electron acceptors, neutral pH and the absence of reduced species
-
II.
Plume core where petroleum hydrocarbons degradation occurs, DIC/alkalinity amounts are high, iron and sulphate reducing conditions and methanogenesis occur accompanied by precipitation of iron and manganese carbonates
-
III.
Plume fringe with steep concentration gradients of electron acceptors; also reducing conditions for nitrate and manganese, and partly iron-reducing conditions accompanied by precipitation of Fe(III) species
-
IV.
Reoxidation of reduced iron and manganese species to phases like ferrihydrite, manganite, cornelite and psilomelane
The precipitation of a mixture of carbonate phases is evident in the most reduced part of the plume. Precipitation of ankerite and kutnahorite is shown to be an important sink of Fe(II) which was released by reductive dissolution of iron oxide and can be deduced from changes in pH and DIC values. The coating of iron oxide surfaces with reduced iron phases in zone II is believed to limit iron reduction and make sulphate reduction and methanogenesis occur in the presence of biologically-available Fe(III). Reoxidation of reduced species in zone IV results in lowering mass fluxes of oxygen and nitrate into the plume and may function as a barrier for penetration of electron acceptors to the plume. Iron-oxide reduction is an important process in this aquifer and this iron cycling may dominate the development of the reducing zones and partly control the size of the plume.
By means of the investigations of the distribution of stable isotopes (δ13C of DIC, and δ18O and δ34S of \( {\text{SO}}_{{\text{4}}} ^{{{\text{2 - }}}} \)), all four zones from the geochemical conceptual model can be confirmed. Background groundwater is in equilibrium with calcite (values δ13C-DIC about −12.7‰ PDB). The reoxidation zone and plume fringe are typified by redox reaction with TPH (δ13C-DIC values ranging from −13.44‰ PDB to −15.58‰ PDB) and a linear trend typified by bacterial sulphate reduction (δ34S increases to a value of 36‰ in the reoxidation zone), and there is significant enrichment in δ13C-DIC by methanogenesis (value −7.6‰).
References
Ahad JME, Lollar BS, Edwards EA, Slater GF, Sleep BE (2000) Carbon isotope fractionation during anaerobic biodegradation of toluene: implications for intrinsic bioremediation. Environ Sci Technol 34:892–896
Bennet PC, Siegel DE, Baedecker MJ, Hult MF (1993) Crude oil in a shallow sand and gravel aquifer. I. Hydrogeology and inorganic geochemistry. Appl Geochem 8:529–549
Bolliger Ch, Schroth MH, Bernasconi SM, Kleikemper J, Zeyer J (2001) Sulphur isotope fractionation during microbial sulphate reduction by toluene-degrading bacteria. Geochim Cosmochim Acta 65(19):3289–3298
Bottrell SH, Hayes PJ, Bannon M, Williams GM (1995) Bacterial sulphate reduction and pyrite formation in a polluted sand aquifer. Geomicrobiol J 13:75–90
Chambers LA, Trudinger PA (1979) Microbiological fractionation of stable sulphur isotopes: a review and critique. Geomicrobiol J 1:249–293
Chapelle FH (1999) Bioremediation of petroleum hydrocarbon-contaminated groundwater: the perspectives of history and hydrology. Ground Water 37:122–132
Christensen TH, Bjerg PL, Banwart SA, Jakobsen R, Heron G, Albrechtsen HJ (2000) Characterization of redox conditions in groundwater contaminant plumes. J Contam Hydrol 45:165–241
Christensen TH, Kjeldsen P, Bjerg PL, Jensen DL, Christensen JB, Baun A, Albrechtsen HJ, Heron G (2001) Biogeochemistry of landfill leachate plumes. Appl Geochem 16:659–718
Cozzarelli IM, Herman JS, Baedecker MJ, Fischer JM (1999) Geochemical heterogeneity of a gasoline-contaminated aquifer. J Contam Hydrol 40:261–284
Cozzarelli IM, Bekins BA, Baedecker MJ, Aiken GR, Eganhouse RP, Tucillo ME (2001) Progression of natural attenuation processes at a crude-oil spill site. I. Geochemical evolution of the plume. J Contam Hydrol 53:369–385
Eganhouse RP, Baedecker MJ, Cozzarelli IM, Aiken GR, Thorn KA, Dorsey TF (1993) Crude oil in a shallow sand and gravel aquifer. II. Organic geochemistry. Appl Geochem 8:551–567
Hackley KC, Liu CL, Coleman DD (1996) Environmental isotope characteristics of landfill leachates and gases. Ground Water 34(5):827–836
Hazdrová M et al (1980) Vysvětlivky k základní hydrogeologické mapě ČSSR 1:200,000, list 02-Ústí nad Labem [Legend to Hydrogeological Map of Czechoslovak Republic 1:s200,000, sheet 02-Ústí nad Labem], Czech Geological Survey, Prague
Herčík, Herrmann,Valečka (1999) Hydrogeologie č;eské kř;ídové pánve [Hydrogeology of the Bohemian Cretaceous basin]. ČGÚ, Prague
Heron G, Christensen TH (1994) The role of aquifer sediment in controlling redox conditions in polluted aquifer: transport and reactive processes in aquifers. Balkema, Rotterdam
Heron G, Christensen TH (1995) Impact of sediment-bound iro on redox buffering in a landfill leachate polluted aquifer (Vejen, Denmark). Environ Sci Technol 29:187–192
Heron G, Christensen TH, Tjell JC (1994a) Oxidation capacity of aquifer sediments. Environ Sci Technol 28:153–158
Heron G, Crouzet C, Bourg AC, Christensen TH (1994b) Speciation of Fe(II) and Fe(III) in contaminated aquifer sediments using chemical extraction techniques. Environ Sci Technol 28:1698–1705
Heron G, Bjerg PL, Gravesen P, Ludvigsen L, Christensen TH (1998) Geology and sediment geochemistry of a landfill leachate contaminated aquifer (Grinsted, Denmark). J Cont Hydrol 29:301–317
Hornibrook ERC, Longstaffe FJ, Fyfe WS (2000) Evolution of stable carbon isotope compositions for methane and carbon dioxide in freshwater wetlands and other anaerobic environments. Geochim Cosmochim Acta 64(6):1013–1027
Hösl P (2003) Hydrogeologické poměry a kontaminace podzemních vod na lokalitě Hnevice [Description of hydrogeological conditions and hydrocarbon contamination of Hněvice site]. MSc Thesis, Charles University of Prague, The Czech Republic
Lovley DR (1997) Potential for anaerobic bioremediation of BTEX in petroleum-contaminated aquifers. J Industr Microbiol Biotech 18:75–81
Mancini SA, Lacrampe-Couloume G, Jonker H, Van Breukelen BM, Grouen J, Volkering F, Sherwood Lollar B (2002) Hydrogen isotopic enrichment: an indicator of biodegradation at a petroleum hydrocarbon contaminated field site. Environ Sci Technol 36:2464–2470
Meckenstock RU, Morasch B, Warthmann R, Schink B, Annweiler E, Michaelis W, Richnow HH (1999) 13C/12C isotope fractionation of aromatic hydrocarbons during microbial degradation. Environ Microbiol 1:409–412
Meckenstock RU, Morasch B, Griebler C, Richnow HH (2004) Stable isotope fractionation analysis as a tool to monitor biodegradation in contaminated aquifers. J Contam Hydrol 75:215–255
Parkhust DL, Appelo CAJ (1999) User’s guide to PHREEQC (Version 2): a computer program for speciation, batch-reaction, one-dimensional transport and inverse geochemical calculations. US Geol Surv Water Resour Invest Rep 99-4259
Postma D, Jakobsen R (1996) Redox zonation: equilibrium constraints on the \( {{\text{Fe}}{\left( {{\text{III}}} \right)}} \mathord{\left/ {\vphantom {{{\text{Fe}}{\left( {{\text{III}}} \right)}} {{\text{SO}}_{{\text{4}}} ^{{{\text{2 - }}}} }}} \right. \kern-\nulldelimiterspace} {{\text{SO}}_{{\text{4}}} ^{{{\text{2 - }}}} } \) reduction interface. Geochim Cosmochim Acta 60(17):3169–3175
Richnow HH, Meckenstock RU, Reitzel LA, Baun A, Ledin A, Christensen TH (2003) In situ biodegradation determined by carbon isotope fractionation of aromatic hydrocarbons in an anaerobic landfill leachate plume (Vejen, Denmark). J Contam Hydrol 64:59–72
Schroth MH, Kleikemper J, Bollinger C, Bernasconi SM, Zeyer J (2001) In situ assessment of microbial sulphate reduction in a petroleum-contaminated aquifer using push-pull tests and stable sulphur isotope analyses. J Cont Hydrol 51:179–195
Spence MJ, Bottrell SH, Thornton SF, Lerner DN (2001) Isotopic modelling of the significance of bacterial sulphate reduction for phenol attenuation in a contaminated aquifer. J Contam Hydrol 53(3–4):285–304
Strebel O, Bőttcher J, Fritz P (1990) Use of isotope fractionation of sulphate-sulphur and sulphate oxygen to asses bacterial desulfurication in a sandy aquifer. J Hydrol 121(1–4):155–172
Tucillo ME, Cozzarelli IM, Herman JS (1999) Iron reduction in the sediments of a hydrocarbon-contaminated aquifer. Appl Geochem 14:655–667
Van Breukelen BM, Rőling WFM, Groen J, Griffioen J, Verseveld HW (2003) Biogeochemistry and isotope geochemistry of a landfill leachate plume. J Contam Hydrol 65:245–268
Wiedemeier TH, Rifai HS, Newell CJ, Wilson JT (1999) Natural attenuation of fuels and chlorinated solvents in the subsurface. Wiley, New York
Wilkes H, Boreham C, Harms G, Zengler K, Rabus R (2000) Anaerobic degradation and carbon isotopic fractionation of alkylbenzenes in crude oil by sulphate-reducing bacteria. Org Geochem 31:101–115
Acknowledgements
This research work was supported by the research grant CORONA (http://www.shef.ac.uk/corona) under the Fifth Framework Programme of the European Commission. This report/article is the sole responsibility of the authors and does not represent the opinion of the Community, nor is the Community responsible for any use and interpretation of the data appearing herein.
The Technical University of Denmark supported the extraction part of the laboratory work. Sulphate isotopic analyses were performed by the laboratories of Queen’s University, Belfast, Northern Ireland. Technical support of the project by OPV Ltd. (Bělohorská 31/264, Prague, Czech Republic) is gratefully acknowledged. This research is being supported by the research grant of the Czech Ministry of Education No. 0021620855 (in review). We would like to thank Z. Vencelides, J.Cizek and F. Buzek for their valuable discussions and Prof. M. Schirmer, Dr. Robert Schneider and anonymous reviewers for their help in improving this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Topinkova, B., Nesetril, K., Datel, J. et al. Geochemical heterogeneity and isotope geochemistry of natural attenuation processes in a gasoline-contaminated aquifer at the Hnevice site, Czech Republic. Hydrogeol J 15, 961–976 (2007). https://doi.org/10.1007/s10040-007-0179-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10040-007-0179-8