
Abstract
The colonial urochordate Botryllus schlosseri is a sedentary species of Mediterranean origin that became cosmopolitan, probably because of postglacial-period dispersal and human-mediated invasions of colonies attached to ship hulls. Here we studied microsatellite allele diversity of Atlantic coast populations from an area ranging from European regions south of the last glacial front to regions that had been permanently ice-covered. Gene diversity levels varied dramatically among populations residing in areas subject to different glacial conditions. Five populations from the Iberian Peninsula, in an area south of the last glacial front, as well as two populations from presumed refugia in Brittany, expressed high gene diversity values (expected heterozygosity [He]: 0.76–0.80; average number of alleles per locus [A]: 7.25–8.75). Two populations inhabiting areas that experienced permafrost conditions (Helgoland Island, Germany, and Plymouth, England) had intermediate values (He: 0.40–0.42; A: 3.0–4.0), whereas the Auchenmalg, Scotland, population, from an area previously covered by ice, showed a remarkably low value (He: 0.17; A: 1.75). Therefore, most European populations of B. schlosseri mirrored the movement of the ice front in the last ice age. A second population from the area that was covered by permanent ice (Lossiemouth, Scotland), however, had a high He of 0.61 and an intermediate A of 3.67. Results were compared with recent invasions (populations less than 200 years old) in the United States and New Zealand that had a higher degree of genetic variation than the European native populations established thousands of years ago. Given the overall dearth of studies on this subject, we suggest that in contemporary established Botryllus populations, gene diversity is affected by ecological factors, some of which can be traced directly to the last ice age. Other parameters of gene diversity are influenced by selection pressure, which might be more intense in northern regions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The global climate has fluctuated widely during the past 3 million years, culminating in the most recent major ice age (Dawson 1992; Hewitt, 1996). These climatic fluctuations have resulted in the latitudinal shift of many organisms (Hänfling and others 2002). In cold periods, most species shifted southward, whereas rapid rises in temperature relocated them northward. Sudden reversals in climate conditions most likely eradicated the vast majority of species over a significant part of their geographical range, leaving small enclaves where populations survived as refugia (Hewitt 1996). Such glacial refuge areas, however, were large enough to harbor a notable fraction of the temperate biota’s intraspecific biodiversity (Petit and others 2003). After an expansion in the postglacial period, intraspecific diversity declined outside the refugia as a consequence of successive founder events (Hewitt 1996, 2000). Species expansion from a leading edge, especially for taxa that are not very mobile, may involve bottleneck events that lead to the loss of alleles and an increase in average population homozygosity (Hewitt 1996). Recent theoretical models have predicted that increased genetic diversity would be achieved mostly through redistribution of the genetic information already present among populations in refugia (Petit and others 2003).
The spatial scale of dispersal and the population structure in the marine environment, are often greater than those documented for terrestrial organisms (Avise 1998). Due to a lack of physical barriers, large population sizes, and high larval dispersal capacities, many marine invertebrate taxa show weak genetic structuring (Palumbi 1996). Nonetheless, several genetic studies on marine organisms have revealed distinct genetic subdivisions, suggesting that high dispersal potential does not necessarily lead to extensive gene flow (see, for example, Palumbi 1996; Avise 1998; Barber and others 2002).
Several recent studies of patterns of postglacial-period expansions, glacial refugia, and population genetic structures have concentrated on marine organisms (Coyer and others 2003; Gysels and others 2004; Provan and others 2005). As these studies have shown, it is often difficult to unequivocally determine the temporal patterns of range expansions or secondary contacts and interactions among populations that have undergone recent fluctuations in population size or distribution (Wares 2002). Therefore, the classical approach of deducing the effects of long-term history on present-day biogeographical distributions is limited, because the actual distribution patterns may be the result of multiple impacts that do not necessarily reflect the assumed history. This is true of cosmopolitan invasive species in which the source populations are diffused (Lambrinos 2004 and literature therein). One such invasive species is the urochordate Botryllus schlosseri.
The cosmopolitan ascidian Botryllus schlosseri, a hard-bottom sedentary marine urochordate species, is globally distributed from subpolar regions to the tropics (Berrill 1950). Colonies are found along the coasts of the Mediterranean Sea and in all European waters up to Norway in the north, in Asia (Korea, Japan, Hong Kong, India), Australia, Tasmania, New Zealand, South Africa, and on the east and the west coasts of the United States (Ruiz and others 2000; Ben-Shlomo and others 2001; Stoner and others 2002; Paz and others 2003; B. Rinkevich unpublished). B. schlosseri colonies are found from intertidal areas to depths of almost 200 m; they are found above and under stones; on algae and seaweed; on pilings, floats, and other artificial substrata; within marinas; and in the wild. This species, most likely of Mediterranean origin (Berrill 1950), became a globally distributed tunicate, probably through continuous introductions and invasions of colonies attached to ship hulls (Berrill 1950; Lambert and Lambert 1998). In contrast to what would be expected given its wide distribution, the microgeographic genetic structure of B. schlosseri is generally consistent with localized gene flow (Grosberg 1987; Yund and O’Neil 2000).
B. schlosseri is an excellent species for studying the relationships among population genetic structure, life history and phylogeography, both for the period extending back to the last glacial maximum (about 20,000 years ago) and for the period of recent invasions (less than 1000 years). Microsatellite investigation of the variability of populations from Israel and Croatia, representing the presumed source of the Mediterranean populations (Rinkevich and others 2001; Paz and others 2003), showed high polymorphism, with an average gene diversity (He) of up to 0.84. Lower yet still substantial gene diversities (He: 0.60–0.69) were recorded in the introduced populations of New Zealand (established less than 150 years ago) and the west and east coasts of the United States (established less than 65 and about 200 years ago, respectively) (Ben-Shlomo and others 2001; Stoner and others 2002).
We studied the genetic diversity of Botryllus schlosseri populations in European sites along the Atlantic coasts, which are specifically relevant to species distribution after the last ice age, from southern Spain and Portugal in the south to Helgoland Island, Germany and Scotland in the north. The analysis considered southern populations that were not subject to frost during the last glacial period, populations from permafrost regions in putative glacial refugia, populations from former permafrost areas, and populations from previously ice-covered regions. A major question was whether the distribution of the former glacial conditions is still mirrored in the observed genetic structures of these populations, or whether recent expansions and invasions of new colonies have blurred the ancient population genetics.
MATERIALS AND METHODS
Sampling
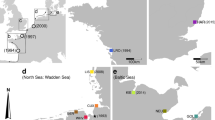
A total of 360 colonies of B. schlosseri from 11 populations were collected from shallow waters along the east coast of the Atlantic Ocean during the summers of 1998–2000 (Table 1 and Figure 1). B. schlosseri colonies are found in natural habitats, as well as within marinas and harbors. Previous work (Paz and others 2003) documented similar levels of gene diversity in colonies collected from manmade habitats, such as an active marina and an old abandoned harbor, as compared to natural rocky habitats. Thus, the type of collecting site is not a limiting factor for comparative analysis. The 11 studied populations represent four different levels for ice-age conditions: five southern populations from areas that were not subject to frost (southern Spain and Portugal), two permafrost populations from putative glacial refugia (Brittany, France), two populations from areas previously covered by permafrost (Plymouth, England, and Helgoland, Germany), and two populations from formerly ice-covered areas (east and west Scotland).

Sampling populations, genetic diversity, and hypothesized conditions under the last ice age. Gene diversity (He) is given in brackets. Numbers of populations are as in Tables 1 and 2. Redrawn from Dawson (1992).
Colonies growing at least 1 m apart from each other were peeled off the substrate and placed in a 1-L container with fresh seawater. We sampled colonies that did not show any morphological signs of natural chimerism (that is, colonies with mixed colors or those characterized by intracolony variations in color morphology) resulting from the fusion of compatible genotypes. DNA extraction and microsatellite typing of colonies followed Ben-Shlomo and others (2001). Genetic variation was assayed using four microsatellites (BS-811, PB-29, PB-41, PB49) (Ben-Shlomo and others 2001). Several samples from different gels and different populations were re-run on the same gel to ensure consistency of scoring between gels runs.
Data Analysis
Allele identification and genotyping were determined directly from the autoradiographs. Sample size differed between populations (Table 2). Collected colonies from each population were removed from the container and numbered at random; for calculation of the average number of alleles/locus (A), we considered the first 20 individuals from each population (in the population from Brest, only 13 colonies were sampled; the population of Sesimbra, which contained only six individuals, was excluded). In populations where numbers of sampling colonies were small, the observed heterozygosity yielded slightly lower values than weighted values; hence, we used the weighted heterozygosity parameter. Weighted observed heterozygosity (Ho), gene diversity (expected heterozygosity, or He) inbreeding coefficient (Fis) and unbiased estimates of Hardy-Weinberg exact P values were computed by the Markov chain method using GENEPOP (Raymond and Rousset 1995), version 3.4 (2003). Previous genetic tests have shown that Botryllus schlosseri populations are characterized by low observed heterozygosity (Ben-Shlomo and others 2001; Stoner and others 2002); thus, for the exact test, H1 was heterozygote deficiency. The significance level was determined after 10,000 dememorizations and 100 batches of 5000 interactions each. The fixation index (Fst) was estimated by a weighted analysis of variance (Weir and Cockerham 1984) for all population pairs. Genetic identity, genetic distance, and its graphical representation (UPGMA) were calculated following Nei (1978), using TFPGA software, version 1.3 (Miller 1997). The significance level for population differentiation (pairwise analysis of all populations; exact tests) (Raymond and Rousset 1995) was determined after 1000 dememorization steps and 20 batches of 2000 permutations per batch, using TFPGA software, version 1.3 (Miller 1997).
RESULTS
The four tested microsatellites were highly polymorphic, revealing 103 different alleles for the 11 sampled populations (11, 25, 22, and 45 for the loci PB-29, PB-41, PB-49, and BS-811, respectively). Allele distributions in every locus at each population are summarized in Figure 2. Observed and expected heterozygosity, the fixation index (Fis) values, and the estimated average number of alleles per locus (for n = 20) are presented in Table 2. Observed heterozygosity was much lower than expected, and Fis values were positive and high (0.39–0.83) in all tested populations. A Hardy-Weinberg exact test for all loci and for all populations revealed a highly significant heterozygote deficiency (P value 0.0000; H0 = HW equilibrium).

The levels of gene diversity (He) and average number of alleles per locus (A n = 20) varied dramatically among populations subject to different conditions in the last ice age (Table 2 and Figure 1). The five southern populations from the Iberian Peninsula, which did not experience frost conditions in the last ice age, expressed high values of He (0.76–0.80) and A (7.25–8.75). Similar values also characterized both Brittany (France) populations, which inhabited a refugium area (Coyer and others 2003). The Helgoland Island (Germany) and Plymouth (England) populations, from areas that experienced permafrost conditions during the last glacial period (Dawson 1992), had intermediate He and A values (0.40–0.42 and 3.00–4.00 respectively), whereas the Auchenmalg, Scotland, population, from an area that was covered by ice, had low values (He: 0.17; A: 1.75). The second Scottish population, Lossiemouth, from an area that was also covered by permanent ice, had—surprisingly—a high He value of (0.61) and an intermediate A (3.67).
Allele frequencies and distribution differed in the tested populations, enabling them to be differentiated (Figure 2). Genic differentiation for each population pair across all loci was highly significant (P < 0.00006, Fisher’s method). The degree of subpopulation structure and differentiation among the populations (Fst, estimated by a “weighted” analysis of variance) (Weir and Cockerham 1984) is presented in Table 3. The differences between the Spanish and the Portuguese populations, as well as between the two French populations, were the lowest (mostly less than 0.07), whereas the differentiation between the Iberian and the French populations was a little higher (0.1). The differentiation between these groups of populations and those of either Helgoland Island or England and Scotland was much higher (more than 0.3). Genetic differences among UK populations and between them and the other populations were relatively high, with the highest differentiation expressed by Auchenmalg in west Scotland (more than 0.5).
The significance level for population differentiation (exact tests) (Raymond and Rousset 1995) was determined by pairwise analysis of all populations. The combined probabilities for each pairwise comparison yielded highly significant differentiation (P = 0.000 for each pair). The differentiation between the populations is depicted by the genetic identity and distance parameters (Nei 1978) and is presented in Table 4 and the resulting tree in Figure 3. Three major nodes (distance, more than 1.5) describe the European Atlantic Ocean populations. One corresponds to the populations of Spain, Portugal, and France; the second represents the population of Helgoland (Germany), which is shown by a separate split, and the third corresponds to the populations of the UK. Within the cluster of the southern populations, the two populations of Brittany (France) are grouped to a separate stem. Interestingly, one Portuguese population (Setubal) is separated from the rest of the Iberian cluster, although it is geographically close to Sesimbra.

Genetic distance between European Atlantic Coast populations. The unrooted phylogenetic tree was based on Nei’s unbiased distance matrix (Nei 1978) and on an UPGMA algorithm.
DISCUSSION
Genetic Diversity, Glacial Gradient, and Latitudinal Gradient
The latitudinal gradient of decreasing biological richness from tropical toward polar regions is well documented across a diverse array of taxa and environments (Willig and others 2003 and literature therein). The same pattern is also reflected in the decline of gene diversity within species (Nevo and others 1984). In addition, populations of plants and animals show lower levels of intraspecific variation in areas severely affected by the Pleistocene glaciations (Taberlet and others 1998; Cronberg 2000). The last warming from full glacial conditions started around 18,000 years bp, culminating in the current warm interglacial climate (Hewitt 1999). After warming, genetic variation in contemporary populations is expected to be lost because of bottleneck effects in geographically restricted refugia and as a consequence of repeated founder episodes (Hewitt 1996). Martin and McKay (2004) compared mtDNA variation among 60 vertebrate species (collectively spanning six continents, two oceans, and 119° latitude) and concluded that seasonality, reduced energy, and the effect of the glacial cycle all have a greater impact on higher-latitude populations.
Therefore, it is expected that genetic diversity across the European continent would be affected by the consequences of the isolation of species in southern refugia during the ice age and by their postglacial colonization northward (Schmitt and Hewitt 2004). Our sampled B. schlosseri populations covered more than 21.5° latitude, ranging from the Iberian Peninsula in the south (Gibraltar, 36.133°N) to Scotland in the north (Lossiemouth, 57.716°N), and thus represent regions that experienced different conditions in the last ice age (Table 1 and Figure 1). During the period of the last glacial maximum, the glacial front was located near the present-day northern coast of the Iberian Peninsula (Frenzel and others 1992; Coyer and others 2003). The Iberian Peninsula was free of ice and Brittany in (France) was on the edge of the permafrost region, a presumed refugium area for marine organisms (Coyer and others 2003). Plymouth (England), and Helgoland Island (Germany) were under permafrost, and Scotland was covered by ice (Dawson 1992). The level of gene diversity (He) (Table 2 and Figure 1) and the average number of alleles per locus (A) (Table 2) of these populations mirrored these conditions, except for one population. The highest He (0.78 ± 0.02) and A (8.2 ± 0.7) values were found in the populations from the Iberian Peninsula and Brittany (0.79 ± 0.03 and 7.5, respectively). The two populations that had been under permafrost showed significantly lower genetic variability values (He: 0.41 ± 0.01 and A: 3.5 ± 0.7); and one of the Scottish populations, Auchenmalg, from an area that was permanently covered by ice, had very low genetic diversity (He: 0.17; A: 1.75). Indeed, the Auchenmalg values were significantly lower than all the other groups. Only one population, from Lossiemouth, Scotland, had a surprisingly high He and an intermediate A.
Lossiemouth is the northernmost sampling point in this study, and the population was collected from a busy marina. The recent anthropogenic introduction of B. schlosseri is a well-documented phenomenon (Berill 1950; Lambert and Lambert 1998; Ben-Shlomo and others 2001; Stoner and others 2002), so it is possible that the genetic structure of the Lossiemouth population was influenced by recent repeated invasions from different sources, leading to higher genetic variation. Indeed, the genetic distance between this population and other European populations was lower relative to the other sampled UK populations (Table 4). Alternatively, it is also possible that although the region was covered by ice, this population originated from an unknown refuge colony. Marine species of northern regions found in the Southern Bight area of the North Sea (Gysels and others 2004) and northeast Iceland (Provan and others 2005) have recently been classified as refuge populations.
Interestingly, although it has suggested that the English Channel and western coast of Ireland provided marine glacial refugia (Provan and others 2005), neither the presence of refuge populations nor the anthropogenic-related movement of individuals among coastline areas affected the variability parameters of the other three northern populations (Plymouth and Auchenmalg in the UK and Helgoland in Germany). The genetic diversity parameters of these populations were low and reflected the conditions extant under the last ice age. The three populations each expressed a common allele (frequency, more than 0.5) (Figure 2) in every tested locus, suggesting predominant founder genotypes. The common genotypes varied between populations (Figure 2), suggesting different founders and limited gene flow between populations. This result is puzzling, because boat movement along the coasts is common. The establishment of each population, and perhaps also its local adaptations, may increase its advantage in terms of dispersing individuals. It is possible that the population of Lossiemouth is not yet fully established and thus has not reached equilibrium; hence, it may be more affected by repeated colonization events.
GENETIC VARIABILITY AND ANTHROPOGENIC INVASION
B. schlosseri is one of the known marine invasive species of the last millennium. Since the beginning of the Age of Exploration, traveling between continents have bridged natural barriers (Mooney and Cleland 2001). Even today, shipping remains the most important vector for biological introductions (Ruiz and others 2000). The spread of exotic species and climatic change are among the most serious threats to the global environment. Although each independently causes considerable ecological damage (Stachowicz and others 2002), the founder effects of invasive populations should be stronger because the time from the founding episode is shorter. It is expected, therefore, that species subject to both processes—climatic changes and recent introductions into new habitats—will express high genetic diversity in well-established old populations, lower variability in populations affected by Pleistocene glaciations, and very low variation in establishing invasive populations.
This is not the case with B. schlosseri, in which very young invading populations that are only several decades old showed higher values of gene diversity than populations established thousands of years before. Relatively high genetic diversity was found in populations of newly established B. schlosseri from New Zealand and the east and west coasts of the United States. Colonization time in New Zealand was estimated at less than 150 years ago, but the gene diversity (He) of the population was 0.55–0.69. In the United States, the east coast population was established 200 years ago, and its He was 0.48–0.66, whereas the west coast population was established 65 years ago, and its He was 0.64–0.74 (Ben-Shlomo and others 2001; Stoner and others 2002). Newly introduced populations may be characterized by high gene diversity resulting from repeated colonizations that increase the gene pool, interactions with the new environmental conditions, and interactions of the newly introduced genotypes with native competitors that may diversify genetic background (Lambrinos 2004). Although sites in the United States may have been repeatedly invaded by new genotypes (Lambert and Lambert 1998), the New Zealand sites were targeted by potentially fewer genotypes and the founder genotypes are still evident (Ben-Shlomo and others 2001). Yet these younger populations, which were less than 150 years old, expressed higher He than the northern European populations that last experienced glacial conditions some 18,000 years ago. These findings suggest that in newly invading populations high genetic diversity may also have resulted from competition with unfamiliar local species, different types of selection, and possibly higher mutation rates.
Differentiation within and between Populations
The effects of the last ice age are also reflected in the allele distribution within and between sampled populations (Figure 2 and Table 2). Mutations at microsatellite loci tend to result in alleles with number of repeat units close to those of the alleles from which they were derived (Goldstein and others 1995). The difference in repeat score between alleles reflects the time that passed between ancestral and the present portrait (Goldstein and others 1995). Therefore, long-established populations are expected to express higher numbers of alleles at low frequency and a wider distribution of sizes. This pattern was found in the southern populations resident in the Iberian Peninsula and Brittany (Figure 2 and Table 2). The frequencies of each allele in these populations were similar—that is, there was no predominant allele. In the northern populations that were subject to permafrost or were covered by ice (Helgoland and the UK sites), however, founder alleles that showed higher frequencies were evident.
Positive Fis values resulting from a high inbreeding rate were found in all our European populations, in accordance with previously studied Botryllus schlosseri populations (Ben-Shlomo and others 2001; Stoner and others 2002; Paz and others 2003). This population attribute, which characterizes all Botryllus populations, probably results from the combined effects of nonrandom larval recruitment (Grosberg and Quinn 1986; R. Ben-Shlomo and B. Rinkevich unpublished) and the subdivision of local populations into spatially isolated populations. Population subdivision resulted from the rapid settlement of the tadpole larvae after a short planktonic phase, within minutes of hatching (Boyd and others 1986). Thus, every local colony harbors private alleles, differentiating it from neighboring populations. This subpopulation structure is best shown by the estimated Fst values, where in most cases geographically close populations expressed lower values, especially those of the Iberian Peninsula and France.
Genic differentiation among populations was highly significant for all loci, in every pair of populations analyzed. This phenomenon is best seen in the comparatively low values of genetic identity (Nei 1978) (Table 4), where the highest identity value between populations was lower than 0.86. Strikingly, identity values between UK populations and Iberian and French populations were extremely low (less than 0.2). Several geographically close populations expressed an unexpectedly low genetic identity. For example, the identity values of two adjacent Portuguese populations, Sesimbra and Setubal, only 30 km apart, was 0.48; and the identity values between Brittany and Plymouth populations (less than 250 km apart) was only 0.04. Because B. schlosseri is the only Botryllus species residing in European waters (Berrill 1950), these low identity values present differences between populations of the same species.
Although the mobile larval stage of B. schlosseri is short-lived, its gene flow is not restricted. During the last millennium, this species became invasive as it was widely dispersed from colonies attached to the hulls of ships (Berrill 1950; Lambert and Lambert 1998). Having spread to both the northern and southern hemispheres, it is now known as a cosmopolitan species. This species, presumably of Mediterranean origin, is found today along both the east and the west coasts of the Atlantic and the Pacific oceans. Populations are found on all continents, except for the polar regions (Ruiz and others 2000; Ben-Shlomo and others 2001; Stoner and others 2002; Paz and others 2003; B. Rinkevich unpublished). Still, the European populations studied here showed remarkable differentiation between geographically adjacent populations and level of genetic diversity that mirrored the relative climatic conditions of the last ice age. These seemingly contradictory findings for B. schlosseri—that an invasive species shows such high genetic differentiation between closely situated populations—suggests local adaptations to different selection pressures. Growing space is a limiting factor for sessile marine invertebrates (Hart and Grosberg 1999). It is therefore possible that a resident B. schlosseri population, once established, either prevents the infiltration of individuals originating from neighboring populations and/or is more successful in the local competition for the acquisition of substrate.
References
Avise JC. 1998. Conservation genetics in the marine realm. J Hered 89:377–82
Barber PH, Palumbi SR, Erdmann MV, Moosa MK. 2002. Sharp genetic breaks among populations of Haptosquilla pulchella (Stomatopoda) indicate limits to larval transport: patterns, causes, and consequences. Molec Ecol 11:659–74
Ben-Shlomo R, Douek J, Rinkevich B. 2001. Heterozygote deficiency and chimerism in remote populations of a colonial ascidian from New Zealand. Mar Ecol Progr Se 209:109–17
Berrill NJ. 1950. The Tunicata. London: Bernard Quaritch
Boyd HC, Brown SK, Harp JA, Weissman IL. 1986. Growth and sexual maturation of laboratory-cultured Monterey Botryllus schloserri. Biol Bull 170:91–109
Coyer JA, Peters AF, Stamand WT, Olsen JL. 2003. Post–ice age recolonization and differentiation of Fucus serratus L. (Phaeophyceae; Fucaceae) populations in northern Europe. Molec Ecol 12:1817–29
Cronberg N. 2000. Genetic diversity of the epiphytic bryophyte Leucodon sciuroides in formerly glaciated versus nonglaciated parts of Europe. Heredity 84:710–20
Dawson AG. 1992. Ice age Earth: late Quaternary geology and climate. London: Routledge
Frenzel B, Pecsiand M, Velichko AA. 1992. Atlas of paleoclimates and paleoenvironments of the Northern Hemisphere: late Pleistocene–Holocene. Budapest: Geographic Research Institute, Hungarian Academy of Science
Goldstein DB, Ruiz Linares A, Cavalli-Sforza LL, Feldman MW. 1995. An evaluation of genetic distances for use with microsatellite loci. Genetics 139:463–71
Grosberg RK. 1987 Limited dispersal and proximity-dependent mating success in the sessile colonial ascidian Botryllus schlosseri. Evolution 41:372–84.
Grosberg RK, Quinn JF. 1986. The genetic control and consequences of kin recognition by the larvae of a colonial marine invertebrate. Nature 322:456–9
Gysels ES, Hellemans B, Pampouli C, Volckaert FAM. 2004. Phylogeography of the common goby, Pomatoschistus microps, with particular emphasis on the colonization of the Mediterranean and the North Sea. Molec Ecol 13:403–17
Hänfling B, Hellemans B, Volckaert FAM, Carvalho GR. 2002. Late glacial history of the cold-adapted freshwater fish Cottus gobio, revealed by microsatellites. Molec Ecol 11:1717–29
Hart MW, Grosberg RK. 1999. Kin interactions in a colonial hydrozoan (Hydractinia symbiolongicarpus): population structure on a mobile landscape. Evolution 53:793–805.
Hewitt GM. 1996. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–76.
Hewitt GM. 1999. Post-glacial re-colonization of European biota. Biol J Linn Soc 68:87–112
Hewitt GM. 2000. The genetic legacy of the Quaternary ice ages. Nature 405:907–13
Lambert CC, Lambert G. 1998. Non-indigenous ascidians in southern California harbors and marinas. Mar Biol 130:675–88
Lambrinos JG. 2004. How interactions between ecology and evolution influence contemporary invasion dynamics. Ecology 85:2061–70
Martin PR, McKay JK. 2004 Latitudinal variation in genetic divergence of populations and the potential for future speciation. Evolution 58:938–45
Miller MP. 1997. Tools for population genetic analyses (TFPGA). Flagstaff (AZ): Department of Biological Sciences, Northern Arizona University
Mooney HA, Cleland EE. 2001. The evolutionary impact of invasive species. Proc Nat Acad Sci USA 98:5446–51
Nei M. 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–90
Nevo E, Beiles A, Ben-Shlomo R. 1984. The evolutionary significance of genetic diversity: ecological, demographic and life history correlates. In: Mani GS, Editor. Evolutionary dynamics of genetic diversity: lecture notes in biomathematics; vol 53: Berlin: Springer-Verlag. p 13–213
Palumbi SR. 1996. Microspatial genetic structure and speciation in marine taxa with high dispersal abilities. In: Ferraris JD, Palumbi SR, Eds. Molecular zoology. New York: Wiley-Liss
Paz G, Douek J, Mo C, Goren M, Rinkevich B. 2003. Genetic structure of Botryllus schlosseri (Tunicata) populations from the Mediterranean coast of Israel. Mar Ecol Progr Ser 250:153–62
Petit RJ, Aguinagalde I, de Beaulieu J-L, Bitlkau C, Brewer S, Cheddadi R, Ennos R, et al. 2003. Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–5.
Provan J, Wattier RA, Maggs CA. 2005. Phylogeographic analysis of the red seaweed Palmari palmate reveals a Pleistocene marine glacial refugium in the English Channel. Molec Ecol 14:793–803
Raymond ML, Rousset F. 1995. An exact test for population differentiation. Evolution 49:1280–3
Rinkevich B, Paz G, Douek J, Ben-Shlomo R. 2001. Allorecognition and microsatellite allele polymorphism of Botryllus schlosseri from the Adriatic Sea. In: Sawada H, Yokosawa H, Lambert CC, editors. The biology of ascidians. Tokyo: Springer. P 426–35
Ruiz GM, Rawlings TK, Dobbs FC, Drake LA, Mullady T, Huqand A, Colwell RR. 2000. Global spread of microorganisms by ships. Nature 408:49–50
Schmitt T, Hewitt GM. 2004. The genetic pattern of population threat and loss: a case study of butterflies. Molec Ecol 13:21–31
Stachowicz JJ, Terwin JR, Whitlatch RB, Osman RW. 2002. Linking climate change and biological invasions: ocean warming facilitates nonindigenous species invasions. Proc Nat’ Acad of Sci U S A 99:15497–500
Stoner DS, Ben-Shlomo R, Rinkevich B, Weisman I. 2002. Genetic Variability of Botryllus schlosseri invasions to the east and west coasts of USA. Mar Ecol Progr Ser 243:93–100
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF. 1998. Comparative phylogeography and postglacial colonization routes in Europe. Molec Ecol 7:453–64
Wares JP,. 2002. Community genetics in the Northwestern Atlantic intertidal.Molec Ecol 11:1131–44
Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38:1358–70
Willig MR, Kaufman DM, Stevens RD. 2003. Latitudinal gradients of biodiversity: pattern, process, scale, and synthesis. Annu Rev Ecol Syst 34:273–309
Yund PO, O’Neil PG. 2000. Microgeographic genetic differentiation in a colonial ascidian (Botryllus schlosseri) population. Mar Biol 137:583–8.
Acknowledgements
This work was supported by a grant from the US–Israel Binational Science Foundation (2003/10), by the Israel Science Foundation (456/01), and by the Marine Genomics NoE. We thank the anonymous referees, whose helpful suggestions contributed significantly to this study. We also thank G. Mass and I. Shaked for helping in animal collection and A. Kerman for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ben-Shlomo, R., Paz, G. & Rinkevich, B. Postglacial-period and Recent Invasions Shape the Population Genetics of Botryllid Ascidians along European Atlantic Coasts. Ecosystems 9, 1118–1127 (2006). https://doi.org/10.1007/s10021-006-0141-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10021-006-0141-y