
Abstract
MicroRNAs (miRNAs) are small noncoding RNAs that take part in diverse biological processes by suppressing target gene expression. Elevated expression of miR-21 has been reported in many types of human cancers. Radiotherapy is a standard adjuvant treatment for patients with glioblastoma. However, the resistance of glioblastoma cells to radiation limits the success of this treatment. In this study, we found that miR-21 expression was upregulated in response to ionizing radiation (IR) in U251 cells, which suggested that miR-21 could be involved in the response of U251 cells to radiation. We showed that a miR-21 inhibitor enhanced IR-induced glioblastoma cell growth arrest and increased the level of apoptosis, which was probably caused by abrogation of the G2-M arrest induced by IR. Further research demonstrated that the miR-21 inhibitor induced the upregulation of Cdc25A. Taken together, these findings suggest that miR-21 inhibitor can increase IR-induced growth arrest and apoptosis in U251 glioblastoma cells, at least in part by abrogating G2-M arrest, and that Cdc25A is a potential target of miR-21.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme is the most common type of all brain tumors and is almost always fatal. Despite the recent advances in therapeutic strategies, such as surgical resection and adjuvant radiotherapy and chemotherapy, the median survival time of glioblastoma multiforme patients is less than 1 year from diagnosis [1]. New classes of treatment modalities, such as molecular target-directed therapies, are urgently needed. Understanding the molecular pathogenesis of glioblastoma may lead to improved conventional or novel therapeutics.
MicroRNAs (miRNAs) are evolutionarily conserved noncoding RNAs, 18 to 26 nucleotides long, that negatively regulate the expression of target genes by interacting with complementary sites in the 3′-untranslated region (UTR) of the gene. They control a diverse range of biological processes, including cell differentiation, proliferation, and apoptosis [2]. One of these microRNAs, miR-21, is overexpressed in human cancers such as breast cancer, liver cancer, pancreatic cancer, colorectal cancer, and glioblastoma [3]. Chan et al. [4] found that miR-21 is an antiapoptotic factor in human glioblastoma cells. Furthermore, miR-21 knockdown disrupts glioma growth and displays synergistic cytotoxicity with S-TRAIL in in vivo xenografts of U87 cells [5]. At present, miR-21 is considered to be a new oncogene that targets several tumor suppressor genes [6]. However, its mechanism of action is largely unknown, and the precise role(s) of miR-21 in cancer remains to be defined.
The G2 checkpoint is a key guardian of the cancer cell genome, and cells depend on the G2 checkpoint for repair of DNA damage. Because abrogation of the G2 checkpoint prevents cells from repairing DNA damage and forces apoptosis, it has emerged as an attractive therapeutic target for anticancer therapies such as radiation [7]. The cell division cycle 25 (Cdc25) family of proteins, which contain highly conserved domains for dual-specificity phosphatases, dephosphorylate and activate cyclin-dependent kinase complexes. Three isoforms, Cdc25A, Cdc25B, and Cdc25C, have been identified in mammalian cells [8]. Damage caused by ionizing radiation and other DNA-damaging agents leads to activation of ataxia-telangiectasia-mutated (ATM) and ATR (ATM and Rad3-related) kinases, which then activate the checkpoint transducer kinases Chk1 and Chk2. The activation of Cdc25A by Chk-1 initiates the multistep process of ubiquitin-mediated proteasomal degradation and leads to cell-cycle arrest. At present, Cdc25A is thought to be the main p53-independent mechanism responsible for cell-cycle arrest or delay in the G1, S, and G2 phases of the cell cycle [9] (additional Fig. 1).
We herein describe our detection of the expression of miR-21 after γ-irradiation. We discuss our investigation of the influence of a combination of IR and an miR-21 inhibitor on the growth and apoptosis of the glioblastoma cell line, U251. We also discuss the possible molecular mechanisms.
Materials and methods
Cell culture
The human glioblastoma U251 cells were maintained in an incubator at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen), and routinely passaged at 2-day intervals. The cells were exposed to IR at room temperature using a Gammacell instrument (Gammacell 40 Exactor, Canada) that can provide a dose rate of 0.81 Gy/min. The cells were exposed for 12 min 21 s to a dose of 10 Gy and for 24 min 42 s to a dose of 20 Gy. The cells were divided into five groups: an untreated group, miR-21 inhibitor group, IR group, IR+ inhibitor negative control group, and IR+ miR-21 inhibitor group. The U251 cells were transfected with a miR-21 inhibitor 48 h before IR treatment.
Trypan blue dye exclusion assay
The viability of cell populations in each group was evaluated by the trypan blue dye exclusion assay. Following all treatments, cells were harvested with trypsin/ethylenediaminetetraacetic acid (EDTA), suspended in phosphate-buffered saline (PBS), and mixed with an equal amount of 0.4% trypan blue stain (Invitrogen). The number of cells excluding trypan blue, representing viable cells, was then counted. The cells were counted in four different fields, and the number of viable cells was calculated as percentage of the total cell population. The count for untreated cells was considered to be 100%.
Isolation of miRNA and real-time RT-PCR
Total RNA was extracted using the miRNeasy Kit (Qiagen). For the SYBR Green-based real-time reverse transcription-polymerase chain reaction (RT-PCR) assays, the miScript PCR System (Qiagen) was used. All primers and probes for the miR-21 and RNU6B endogenous controls for the SYBR green miRNA assays were purchased from Qiagen. The expression of mature miRNAs was determined by real-time PCR, performed according to the manufacturer’s instructions. Relative miRNA expression was calculated via the 2-ΔΔCt method [10].
Transfections
The miR-21 inhibitor and inhibitor negative control were purchased from Qiagen. The cells were transfected using the Lipofectamine 2000 reagent (Invitrogen) when cells were 50% confluent. Transfection complexes prepared according to the manufacturer’s instructions were added directly to the U251 cells at a final oligonucleotide concentration of 75 nmol/l. The transfection medium was replaced 8 h posttransfection.
Caspase-3 and caspase-7 activity
For detection of caspase-3 and caspase-7 activation, the cells were plated on 96-well plates in triplicate wells for each condition, transfected, and exposed to 20 Gy IR 48 h posttransfection. The detection of caspases was performed using the Caspase-Glo 3/7 Assay (Promega) 24 h later according to the manufacturer’s instructions.
Cell-cycle analysis
U251 cells were transfected as described above and exposed to 20 Gy IR for 24 h. The cells were then trypsinized and fixed with ice-cold 70% ethanol overnight at 4°C. After extensive washing, the cells were resuspended in PBS containing 50 μg/ml PI (Sigma-Aldrich) and 50 μg/ml RNaseA (Sigma-Aldrich), incubated for 1 h at room temperature, and analyzed using a FACScalibar instrument (Becton-Dickinson, San Jose, CA, USA). The cell-cycle phase distribution was analyzed with the use of the ModFit LT 3.1 software program (Verity Software House, Topsham, ME, USA).
Western blot analysis
Proteins from each group of cells were extracted with NP40 cell lysis buffer (50 mM Tris, pH 7.4, 250 mM NaCl, 5 mM EDTA, 50 mM NaF, 1 mM Na3VO4, 1% NP40, 0.02% NaN3). One-dimensional sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis was performed with a corresponding gel concentration using the discontinuous buffer system of Laemmli (Bio-Rad Laboratories, Richmond, CA, USA). The electrophoresed proteins were transferred to polyvinylidene difluoride membranes and subjected to an immunoblot analysis with antibodies against Cdc25A (1/200; Santa Cruz Biotechnology, Santa Cruz, CA, USA). The reaction was detected with the enhanced chemiluminescence reagents (Amersham Life Science, Arlington Heights, IL, USA) according to the manufacturer’s protocol. The membranes were reblotted with anti-actin Ab-5 (1:5000; BD Bioscience) after washing to check the equal loading of the gel.
Statistical analysis
All tests were done using the SPSS Graduate Pack 11.0 statistical software package (SPSS, Chicago, IL, USA). Descriptive statistics, including the means and SE, with one-way analysis of variance (ANOVA) and two-way ANOVA, were used to determine whether there were significant differences. P < 0.05 was considered significant.
Results
IR induces upregulation of miR-21 expression in U251cells
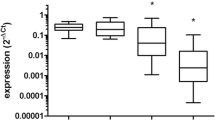
To investigate whether the expression of miR-21 changes after IR, miR-21 expression of U251 cells at different time points after 10 Gy γ-irradiation was analyzed by RT-PCR (Fig. 1). Expression of miR-21 was enhanced at the 3-, 6-, and 12-h time points after 10 Gy γ-irradiation (P < 0.05). The enhanced miR-21 expression in IR-treated U251 cells suggested that miR-21 may play an important role in the response to radiation of U251 cells.

Ionizing radiation (IR)-stimulated microRNA 21 (miR-21) expression in the U251 human glioblastoma cell line. MiR-21 quantitative polymerase chain reaction (PCR) in U251 cells at various time points after IR exposure (10 Gy) showed upregulation of miR-21 after IR treatment (*P < 0.05). Results are described as fold increase compared with nonirradiated U251 cells. Each data point represents the average of three independent experiments
A miR-21 inhibitor augmented IR-induced U251 cell growth arrest
U251 cells were transfected with a miR-21 inhibitor complementary to mature miR-21. RT-PCR analysis revealed that the miR-21 inhibitor effectively reduced the expression level of miR-21 at 48 and 72 h after transfection (Fig. 2a). To examine whether miR-21 knockdown influences IR-induced GBM cell growth arrest, U251 cells were transfected with the miR-21 inhibitor for 48 h and treated with 20 Gy radiation 24 h later. The trypan blue dye exclusion assay was performed to evaluate the U251 cell viability using a hemocytometer 24 h after IR treatment. Treatment with the miR-21 inhibitor led to a decrease in cell viability induced by IR. In addition, compared with the untreated group, a decrease in cell viability was observed in the miR-21 inhibitor group, which is in agreement with a previous report [11] (Fig. 2b).

miR-21 inhibitor augmented IR-induced U251 cell growth arrest. a miR-21 inhibitor reduced the miR-21 level in U251 cells. The histogram shows relative miR-21 expression. A significant difference between the miR-21 inhibitor group (48 and 72 h) and untreated group or control (NC) oligonucleotide group is indicated by *(P < 0.01). b The trypan blue dye exclusion assay assessed the viability of U251 cells. The miR-21 inhibitor augmented the IR-induced decrease in U251 cell viability. Significant differences between the IR group and untreated group or the miR-21 inhibitor group are indicated by *(P < 0.05). Significant differences between the IR+ miR-21 inhibitor group and the IR group or the IR + NC group are indicated by ^(P < 0.05)
The miR-21 inhibitor augmented IR-induced U251 apoptosis
Members of the cysteine aspartic acid-specific protease (caspase) family play key effector roles in apoptosis in mammalian cells, including glioblastoma cells [12, 13]. Activation of caspases-3 and -7 after radiation or miR-21 inhibitor treatment was investigated with a caspase-3/7 activity assay. Cells treated by radiation alone showed a significant increase in caspase-3/7 activation (P < 0.01), and treatment with the miR-21 inhibitor alone also increased caspase-3/7 activation (P < 0.05). However, caspase-3/7 activity in cells treated with radiation + miR-21 inhibitor was increased about 39% (P < 0.01) compared with cells treated with IR alone or with IR+ control (Fig. 3). These results suggested that the miR-21 inhibitor could significantly increase the apoptosis level induced by IR in the U251 glioblastoma cells.

Effects of miR-21 inhibition on caspase-3/7 activity in IR-treated U251 cells. U251 cells were treated with miR-21 inhibitor before IR for 48 h, followed by 20 Gy γ-irradiation for 24 h. Caspase-3/7 activity was measured by the Caspase-Glo 3/7 assay kit. The miR-21 inhibitor group showed an increase in caspase 3/7 activity compared with the untreated group (*P < 0.05). Compared with untreated and negative control groups, an increase in caspase-3/7 activity was observed in the IR group (**P < 0.01); the IR+ miR-21 inhibitor group showed increased caspase-3/7 activity compared to both IR and IR + NC groups (^^ P < 0.01). Data are means of triplicate experiments
The miR-21 inhibitor abrogates IR-induced G2-M arrest in U251 cells by increasing CD25A
DNA damage induced by IR always causes G2-M arrest, and this G2-M arrest may be involved in the radiosensitivity of the cells. We assessed the effects of the miR-21 inhibitor on radiotherapy (RT)-induced G2-M arrest. U251 cells transfected with the miR-21 inhibitor (48 h) were treated with γ-radiation (20 Gy); cells were then incubated for an additional 24 h and harvested for flow cytometry (FCM) analysis. As reported previously [14], U251 cells exposed to IR exhibited G2-M arrest. We found that the miR-21 inhibitor significantly inhibited the IR-induced G2-M arrest of U251 cells (Fig. 4a).

The miR-21 inhibitor abrogated IR-induced G2-M arrest in U251 cells and increased Cdc25A expression. a U251 cells were transfected with miR-21 inhibitor 48 h before exposure to 20 Gy γ-irradiation. After 24 h, cells were harvested for cell-cycle analysis by FACS. IR induced a significant increase in the number of U251 cells in the G2/M phase (**P < 0.01). G2-M arrest was partly reversed in the IR + miR-21 inhibitor group compared with IR and IR + NC groups (^^ P < 0.01). b Cell treatments as described in a. Expression of Cdc25A in each group of cells was detected by Western blot analysis. Increase in Cdc25A expression was observed in the IR + miR-21 inhibitor group compared with the IR and IR + NC groups. Cdc25A expression was also increased in the miR-21 inhibitor group, compared with the untreated group. Data are means of triplicate experiments
Cdc25A has been shown to regulate the G2-M transition, and its downregulation is involved in establishing the G2-M checkpoint following γ-irradiation [8, 15]. To determine whether Cdc25A is involved in the abrogation of the IR-induced G2-M arrest by the miR-21 inhibitor, we evaluated the Cdc25A expression levels of each group. The Cdc25A level decreased following radiation in both the blank control group and negative control cells compared with untreated cells, but the Cdc25A level was increased in the IR + miR-21 inhibitor cells. Furthermore, an increase in the Cdc25A level was also noted in the miR-21 inhibitor group compared to the untreated group (Fig. 4b). These results suggest that miR-21 participates in IR-induced glioma cell-cycle modulation either directly or indirectly through targeting Cdc25A.
Discussion
Radiotherapy is a standard adjuvant treatment for patients with glioblastoma. However, the resistance of glioblastoma cells to irradiation limits the success of this treatment [16]. MiR-21, which is significantly augmented in glioblastoma, has been demonstrated to be an important oncogene that targets several tumor suppressor genes and regulates tumor cell proliferation, apoptosis, and invasion [6]. However, the relationship between irradiation and miR-21 in glioblastoma has not yet been elucidated.
In this study, we first exposed cells to γ-irradiation and investigated the level of miR21 expression. We observed the expression of miR-21 was enhanced after this treatment in U251 cells. Therefore, we hypothesized that miR-21 is likely to be involved in the radiation response of U251 cells and that it might influence the resistance of U251 cells to radiation. After confirming this hypothesis, we showed that the miR-21 inhibitor enhanced RT-induced GBM cell growth arrest and apoptosis level in the U251 cells. Also, the miR-21 inhibitor abrogated IR-induced G2-M arrest in these cells, and we demonstrated that modulation of Cdc25A expression appears to be involved in the molecular mechanism underlying this effect.
The expression patterns of miRNAs that are influenced by cancer treatments, such as chemotherapy or radiotherapy, are increasingly being recognized. The expression of miRNAs may differ depending on cell type, postradiation time, and radiation dose in normal endothelial cells and tumor cells [17–20]. MiR21, which has been demonstrated to be an oncogene, had been reported to be upregulated after irradiation in various cell types [20–22]. In this study, we found that miR-21 expression was upregulated in response to irradiation. These results led us to surmise that miR-21 was involved in the response to radiation, including apoptosis and cell-cycle arrest.
According to the previous reports, G2-M arrest plays a protective role in cells in response to DNA-damaging agents, including temozolomide (TMZ)-induced cytotoxicity, and abrogation of the G2 checkpoint sensitized cells to TMZ [23, 24]. Morgan et al. [25] recently showed that abrogation of the G2 checkpoint and inhibition of homologous recombination repair contributed to radiosensitization in pancreatic cancer. Furthermore, inhibiting the constitutive activation of cell-cycle checkpoints and reversing the cell-cycle blockade had been demonstrated to sensitize CD133+ glioma stem cells to IR and chemotherapy [26]. Our present data showed that miR21 knockdown augmented RT-induced GBM cell growth arrest and apoptosis. Interestingly, we observed that the miR-21 inhibitor abrogated IR-induced G2-M arrest. These results may support the view that abrogation of the checkpoint by the miR21 inhibitor is associated with an increase in the number of cells that die because of premature entry into mitosis. MiR-21 appears to target multiple tumor-suppressive pathways, and inhibition of miR-21 has been observed to induce proliferation arrest and apoptosis in glioblastoma cells [6, 22]. In this study, we also showed that a miR-21 inhibitor induced cell growth arrest and apoptosis, leading to increased radiosensitivity.
Recent studies showed convincing evidence that Cdc25A is a critical regulator of the cell-cycle checkpoint in response to DNA damage caused by IR, oxidative stress, and other DNA-damaging agents [9]. Wang et al. [27] demonstrated that microRNA-21 negatively regulated Cdc25A and cell-cycle progression in colon cancer cells. In addition, Cdc25A was found to be a target of miR-16 and the response to UV-induced DNA damage [21]. Our data indicated that miR-21 knockdown increased the Cdc25A level. These observations may indicate that the important cell-cycle regulator, Cdc25A, is subject to modulation by miRNAs, although the Cdc25A activities are tightly regulated by multiple mechanisms during cell-cycle progression [8]. On the other hand, as U251 cells are p53 mutant cells, the regulation of Cdc25A by miR21 seems to be independent of tumor suppressor p53.
In summary, we have herein shown that miR21 expression increased in response to IR in U251 cells, and that miR21 knockdown augmented RT-induced GBM cell growth arrest and apoptosis. Furthermore, such an effect may be partly caused by a reduction in G2-M cell-cycle accumulation as a result of direct or indirect modulation of the Cdc25A level.
References
Sathornsumetee S, Rich JN (2006) New treatment strategies for malignant gliomas. Expert Rev Anticancer Ther 6:1087–1104
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Cho WC (2007) OncomiRs: the discovery and progress of microRNAs in cancers. Mol Cancer 6:60
Chan JA, Krichevsky AM, Kosik KS (2005) MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 65:6029–6033
Corsten MF, Miranda R, Kasmieh R, Krichevsky AM, Weissleder R, Shah K (2007) MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res 67:8994–9000
Papagiannakopoulos T, Shapiro A, Kosik KS (2008) MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res 68(19):8164–8172
Bucher N, Britten CD (2008) G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer 98:523–528
Boutros R, Dozier C, Ducommun B (2006) The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol 18:185–191
Ray D, Kiyokawa H (2008) CDC25A phosphatase: a rate-limiting oncogene that determines genomic stability. Cancer Res 68:1251–1253
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods 25:402–408
Zhou X, Ren Y, Moore L, Mei M, You Y, Xu P, Wang B, Wang G, Jia Z, Pu P, Zhang W, Kang C (2010) Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab Invest 90:144–155
Kobayashi T, Masumoto J, Tada T, Nomiyama T, Hongo K, Nakayama J (2007) Prognostic significance of the immunohistochemical staining of cleaved caspase-3, an activated form of caspase-3, in gliomas. Clin Cancer Res 13:3868–3874
Bayascas JR, Yuste VJ, Benito E, Garcia-Fernandez J, Comella JX (2002) Isolation of AmphiCASP-3/7, an ancestral caspase from amphioxus (Branchiostoma floridae). Evolutionary considerations for vertebrate caspases. Cell Death Differ 9:1078–1089
Tsuboi K, Moritake T, Tsuchida Y, Tokuuye K, Matsumura A, Ando K (2007) Cell cycle checkpoint and apoptosis induction in glioblastoma cells and fibroblasts irradiated with carbon beam. J Radiat Res (Tokyo) 48:317–325
Mailand N, Podtelejnikov AV, Groth A, Mann M, Bartek J, Lukas J (2002) Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J 21:5911–5920
Chang JE, Khuntia D, Robins HI, Mehta MP (2007) Radiotherapy and radiosensitizers in the treatment of glioblastoma multiforme. Clin Adv Hematol Oncol 5:894–902, 907–915
Maes OC, An J, Sarojini H, Wu H, Wang E (2008) Changes in MicroRNA expression patterns in human fibroblasts after low-LET radiation. J Cell Biochem 105:824–834
Weidhaas JB, Babar I, Nallur SM, Trang P, Roush S, Boehm M, Gillespie E, Slack FJ (2007) MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res 67:11111–11116
Ishii H, Saito T (2006) Radiation-induced response of micro RNA expression in murine embryonic stem cells. Med Chem 2:555–563
Wagner-Ecker M, Schwager C, Wirkner U, Abdollahi A, Huber PE (2010) MicroRNA expression after ionizing radiation in human endothelial cells. Radiat Oncol 5:25
Pothof J, Verkaik NS, van IW, Wiemer EA, Ta VT, van der Horst GT, Jaspers NG, van Gent DC, Hoeijmakers JH, Persengiev SP (2009) MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. EMBO J 28:2090–2099
Zhu Y, Yu X, Fu H, Wang H, Wang P, Zheng X, Wang Y (2010) MicroRNA-21 is involved in ionizing radiation-promoted liver carcinogenesis. Int J Clin Exp Med 3:211–222
Hirose Y, Berger MS, Pieper RO (2001) Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res 61:5843–5849
Tentori L, Portarena I, Torino F, Scerrati M, Navarra P, Graziani G (2002) Poly(ADP-ribose) polymerase inhibitor increases growth inhibition and reduces G(2)/M cell accumulation induced by temozolomide in malignant glioma cells. Glia 40:44–54
Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, Arumugarajah S, Hylander-Gans L, Morosini D, Simeone DM, Canman CE, Normolle DP, Zabludoff SD, Maybaum J, Lawrence TS (2010) Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res 70:4972–4981
Frosina G (2009) DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol Cancer Res 7:989–999
Wang P, Zou F, Zhang X, Li H, Dulak A, Tomko RJ Jr, Lazo JS, Wang Z, Zhang L, Yu J (2009) microRNA-21 negatively regulates Cdc25A and cell cycle progression in colon cancer cells. Cancer Res 69:8157–8165
Acknowledgments
The authors thank Dr. Runze Xun and Dr. Jingshan Lu for their excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Y. Li and S. Zhao contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10014_2011_37_MOESM1_ESM.tif
Additional Fig. 1. When DNA is damaged by radiation (IR), CHK1 is activated, and CHK1 then targets cell division cycle 25A (Cdc25A) to ubiquitin-mediated degradation. The CHK1-dependent degradation of Cdc25A is a major p53-independent mechanism of either cell-cycle arrest or a delay in the G2-M phase. Eventually, DNA damage results in cell-cycle arrest at G2-M. The G2 checkpoint has a protective function; therefore, the fate of the injured cells is determined by the extent of DNA damage and the duration of G2-M. The restored cells thereafter survive, whereas the unrestored cells undergo apoptosis (TIFF 38590 kb)
Rights and permissions
About this article
Cite this article
Li, Y., Zhao, S., Zhen, Y. et al. A miR-21 inhibitor enhances apoptosis and reduces G2-M accumulation induced by ionizing radiation in human glioblastoma U251 cells. Brain Tumor Pathol 28, 209–214 (2011). https://doi.org/10.1007/s10014-011-0037-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10014-011-0037-1