
Abstract
A hybrid nanomaterial with palladium nanoparticles (PdNps) supported on reduced graphene oxide (rGO) flakes, dispersed in an aqueous solution was synthesized through a reproducible and scalable electrochemical method. The rGO–PdNps hybrid with excellent stability and a narrow range of size distribution in the aqueous dispersion is obtained. In a second step, additional plasmonic characteristics were provided by forming Au–Pd bimetallic nanostructures in the rGO flakes, through the gold nanoparticles formation by galvanic replacement on the Pd surface sites at the rGO–PdNps hybrid. The interaction of the rGO–AuPdNps with the PVP binder in aqueous dispersion was investigated by SERS (surface enhancement of Raman scattering) spectroscopy. Furthermore, both rGO–PdNps and rGO–AuPdNps hybrid nanomaterials were deposited by drop-casting on glassy carbon electrodes in order to evaluate their electrocatalytic activity for hydrogen evolution reaction (HER). Experimental findings from cyclic voltammetry in 0.5 M H2SO4 solution show that both hybrids exhibit activation of HER kinetics. Electrodes modified with rGO–AuPdNps show a current density of HER at −0.6 V vs. SCE of 9.71 mAcm−2, while that recorded for glassy carbon electrodes containing rGO was 0.18 mAcm−2 considering the geometrical area of the electrodes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In recent years, intense work has been carried out to find highly controlled and efficient methods to obtain stable aqueous dispersions of nanoparticles [1,2,3,4]. Depending on their applications, some requirements such as particular optical or electrical properties for nanoparticles have to be addressed. Nowadays, innumerable simple metal nanostructures of varied chemical compositions can be obtained by different methods; however, hybrid nanomaterials with graphene derivatives are increasingly necessary to broaden the spectrum of applications. For that, the synthesis methods to be used must be suitable for obtaining highly stable hybrid nanostructures under different configurations such as dispersed in solution [2, 5] or supported on solid platforms [3], also considering the possibility of its production on a larger scale [6].
A wide variety of two-dimensional (2D) nanomaterials has been used for applications in catalysis, mainly due to their unique chemical, physical, and electronic properties. Graphene films have been considered exceptional platforms for particle immobilization in electrocatalysis studies, due to the large available surface area, high stability under ambient conditions, and raised electrical conductivity [7]. In addition, thin flakes of graphene oxide (GO), MoS2, and WS2 have been also used as support materials for diverse metallic nanoparticles [8,9,10,11,12]. GO flakes are often used as microplatforms of Nps because one of their most outstanding characteristics, given by the presence of oxygenated groups, can be modified resulting in layered structures similar to graphene, named reduced graphene oxide (rGO). Numerous protocols with different methods have been used to attain the complete reduction of GO sheets, to approach the structure of pristine graphene [13,14,15,16]. However, residual functional groups and surface defects remain in the structure of the two-dimensional material and severely alter the physicochemical properties of the carbon sheet [9]. Due to these particular characteristics of rGO flakes, the residual functional groups facilitate the interaction with nanoparticles, molecules, and different species, as it has been widely demonstrated as optimal nanomaterials for charge storage, detection of adsorbed molecules, and catalysis among other processes [5].
On the other side, it is well-known that hydrogen is considered one of the most promising green energy vectors since it can be obtained from renewable energy sources such as wind or solar-based technologies [17]. Hydrogen production from water electrolysis has attracted considerable attention due to the high purity fuel obtained [18]. In electrochemical studies, the hydrogen evolution reaction (HER) in an acid medium usually requires the use of catalyst electrodes composed of noble metals. In recent years, many investigations have been carried out by using different nanostructures with a low content of these expensive metals, which are supported on electrodes normally derived from carbon [19]. Thereby, the composition and structure of the electrode material play an important role in its catalytic activity. For example, particles of Pd have been presented as an acceptable substitute catalyst of Pt for HER because of its lower price, excellent catalytic capability, and hydrogen storage [19, 20].
In the literature of recent years, it is found that the graphene-based hybrid materials obtained either by massive metal encapsulation or by deposition of nanoparticles on two-dimensional flakes are particularly important for efficient electrocatalytic activity [6, 21,22,23], without forgetting that it is advisable to obtain a homogeneous distribution of Nps on the 2D material, to be able to act as a composite with great stability, reliability, and high durability. Furthermore, if the synthesis of Nps or hybrids is carried out with reducing agents, some extra complications may come from by-products of the reaction that can contaminate the composite.
On the basis of the last research results, where the use of different supported nanostructures as a nanomaterial composite is widely proposed. Due to the granted effectiveness given by the synergy between the properties of the components, we propose a simple synthesis method (electrochemical first and then, by galvanic replacement) to obtain hybrids with Nps and layered 2D nanomaterials. Thus, the procedures for obtaining supported-metal nanoparticles are not only dependent on the characteristics of the hybrid sought but also specific on its functionality (i.e., electrocatalytic activity). For this reason, although the experimental procedure is more complex than direct chemical synthesis, the electrochemical methods can be used for obtaining 2D composites with low contamination, such as electrochemical deposition [22, 24, 25], spontaneous deposition [26], galvanic replacement approach [27], and chemical reduction [21, 28].
In this work, we present a simple approach to preparing rGO–AuPdNps hybrid nanomaterials. The experimental procedure combines the electrochemical synthesis of Pd nanoparticles supported on reduced graphene oxide flakes (rGO) in an aqueous solution with subsequent Au decoration on the PdNps surface. The last step is reached by the addition of HAuCl4 to the ink of rGO–PdNps for the rGO–AuPdNps hybrid to be formed by the galvanic replacement reaction that occurs between Au and Pd. Properties of resulting hybrid materials, such as particle distribution, structure, surface morphology, and chemical composition were determined by XRD, TEM, and SEM–EDS methods. The electrocatalytic activity for hydrogen evolution reaction of electrodes after drop-casting of rGO–Nps hybrids ink was very noticeable. Kinetic parameters and electrocatalytic efficiency for HER in 0.5 M H2SO4 were compared with different two-dimensional structured materials, such as graphene and MoS2 flakes. In addition, based on the plasmonic properties of the rGO–AuPdNps hybrid, surface enhancement of Raman spectroscopy (SERS) was used to confirm the presence of polyvinylpyrrolidone (PVP) stabilizer molecules, with a very sensitive record. This result was supported by a theoretical analysis of the electric field enhancement, promoted by the AuPdNps bimetallic nanostructures which were performed with electrodynamic calculations. The procedure for the synthesis of rGO–Nps hybrids with bimetallic particles provides an effective method for the production of stable metal Nps in an aqueous solution at a large scale.
Experimental Section
Materials
Palladium (II) chloride PdCl2 (tetrahedron) dissolved in 0.5 M HCl (Cicarelli), chloroauric acid trihydrate HAuCl4.3H2O (Sigma-Aldrich), 4 mg/mL graphene oxide GO aqueous dispersion (GO) (Graphenea), H2SO4 (Cicarelli), KNO3 (Cicarelli), and polyvinylpyrrolidone polymer (Mw PVP = 10,000 Da) (Sigma-Aldrich) were used as received. Solutions were prepared with deionized water (resistivity ≈ 18 MΩcm−1) from a Milli-Q Millipore system and deoxygenated with high-purity nitrogen before each electrochemical experiment.
Synthesis of rGO–Nps hybrids
The synthesis of rGO–PdNps hybrids was carried out in a conventional three-electrodes electrochemical cell, employing as electrolyte an aqueous solution of KNO3 (0.1 M), H2PdCl4 (0.5 mM), PVP (16 g L−1), and 2.5 mL of 0.08 g L−1 aqueous dispersion of GO flakes in a final volume of 40.0 mL. In order to obtain the hybrid nanomaterial, the commercial aqueous dispersion of GO was diluted before addition to the electrolyte, resulting in a final concentration of 0.005 g L−1.
The working electrode was a polycrystalline gold rotating disc electrode (RDE) (0.196 cm2 area), while a large area Pt sheet and a saturated calomel electrode (SCE) were used as the auxiliary and reference electrodes, respectively. Electroreduction of H2PdCl4 and GO species at the interface gold electrode/electrolyte was effectively performed by a galvanostatic pulse, with strong agitation of solution. Finally, the incorporation of gold into the hybrids was carried out by the addition of different aliquots of 24 mM HAuCl4 solution. Thus, the rGO–AuPdNps dispersion was obtained by spontaneous Au nucleation on the PdNps surface.
Characterization of the synthesized nanomaterials
As synthesized, rGO–PdNps and rGO–AuPdNps hybrids were characterized by transmission electron microscopy (TEM). Pretreatment of the dispersions was required for the correct characterization and evaluation of the properties of the hybrid nanomaterial. The aqueous dispersions were centrifuged (at 6000 rpm) and rinsed with water, three times in successive steps, in order to eliminate the residues not incorporated in the rGO flakes. For microscopic characterization, samples were prepared by deposition of one drop (∼ 10 μL) of pretreated dispersion onto a holey carbon-formvar-coated copper TEM grid (100 mesh from Ted Pella Inc.). TEM images of particles were obtained using a JEM-JEOL 1120 EXII transmission electron microscope, working at 80 keV. The mean diameter of Nps was determined by using Image J software. For elemental analysis, pretreated dispersions were drop-casting deposited on a silicon wafer. A Supra 40 microscope (Zeiss Company) FESEM operating at 3–6 kV equipped with an EDS system operating at 6–15 kV was used.
Crystalline structure characterization of rGO–PdNps and rGO–AuPdNps nanomaterials was performed by X-ray diffraction (XRD) using a PANalytical X’Pert PRO diffractometer (40 kV, 40 mA) in the θ–2θ Bragg–Brentano geometry at room temperature. A 2θ range was selected between 10° and 70°, with steps of 0.02° and an integration time of 14 s per step.
The XPS data were collected using Al Kα radiation at 1486.69 eV (150 W, 10 mA), a charge neutralizer, and a delay-line detector (DLD) consisting of three multi-channel plates. Binding energies are referred to as the C1s peak at 285 eV. Survey spectra were recorded from −5 to 1350 eV at a pass energy value of 150 eV (number of sweeps: 2) using an energy step size of 1 eV and a dwell time of 100 ms. High-resolution spectra for Pd3d and Au4f were recorded in the appropriate regions, at a pass energy value of 20 eV using an energy step size of 0.05 eV and 0.1 eV respectively. The spot size was 400 μm.
Optical characteristics of rGO–Nps dispersions were recorded by UV–vis absorption spectroscopy in the range of 300–1100 nm. Extinction spectra were recorded with a Shimadzu UV–1601 spectrophotometer, using a quartz cell with a 1.0 cm path length at room temperature.
Stability of the hybrid nanomaterial
The stability of the nanomaterial in the aqueous dispersion is given by the PVP molecules, which are present in the electrochemical synthesis. In order to characterize the hybrid nanomaterial, the presence of the PVP stabilizing agent covering it was analyzed. For this, Raman spectra of freshly synthesized rGO–AuPdNps dispersion, without any further treatment, have been measured taking advantage of the SERS activity given by the hybrids themselves. This evaluation was performed by recording Raman spectra from the aqueous dispersion containing particles synthesized with major content of gold. Spectroscopic data were obtained using a Horiba Jobin–Yvon Raman microscope (LABRAM–HR 800), with a 4 × objective lens and 632.8 nm laser (3.5 mW) with a spectral resolution of 1.5 cm−1. Additional electrodynamic calculations with the optical response modeling for rGO–AuPdNps hybrids were performed using Lumerical FDTD solutions software. The structural dimensions were based on the TEM analysis. In all calculations, the dielectric functions were taken from Johnson and Christy [29] in the case of gold, and palladium tabulated by Palik [30]. Furthermore, the refractive index of the carbonaceous material was taken from Weber et al. [31]. The background media (aqueous) was assumed to have a refractive index of 1.33.
Evaluation of the electrocatalytic activity for HER
Analysis of the electrochemical behavior of electrodes containing rGO and rGO–Nps deposited by dispersion drop-casting (50 μL) on glassy carbon was carried out in a conventional three-electrode cell at room temperature. A glassy carbon disc (0.070 cm2), a large area platinum sheet, and a saturated calomel electrode SCE (0.243 V vs. RHE) were employed as working, auxiliary, and reference electrodes, respectively.
In order to establish the active surface area of nanoparticles, PdNPs or AuPdNps included in the carbon network of rGO supported onto the glassy carbon electrodes, potentiodynamic experiments of Cu underpotential deposition (UPD) in 5.0 mM CuSO4 and 0.5 M H2SO4 solution were performed.
For the evaluation of the electrocatalytic activity of the nanomaterials, deoxygenated 0.5 M H2SO4 solution was used for potentiodynamic scans in the potential range of HER, performed with an Autolab potentiostat/galvanostat model PGStat30 with FRA2 module. The ohmic drop correction (~ 50 Ω) was automatically performed by Autolab software (Metrohm Autolab Nova 1.10). In the voltammetric experiments, the potential was scanned between 0.1 and −0.6 V (vs. SCE) at 0.01 V·s−1 for all electrodes.
Results and discussion
Electrochemical synthesis of the rGO–PdNps aqueous dispersion
The aqueous dispersion of the rGO–PdNps hybrid was obtained by simultaneous electroreduction processes of GO flakes and PdCl42− ions. A constant current density pulse of −100 mA cm−2 for 10 min to the gold rotating disc electrode at 1000 rpm was performed in deoxygenated KNO3 electrolyte containing the electroactive species. Under these conditions, the electrode potential evolves reaching significant negative values, where the reduction of both, PdCl42− ions and GO flakes occurs [32]. In addition, other processes take place simultaneously with the formation of the hybrid nanomaterial. Thus, rGO flakes play the role of support platforms for Nps, by their incorporation in the carbon matrix, which is integrally stabilized in solution with the PVP molecules [33]. Similarly, a dispersion of PdNps was obtained and evaluated as an efficient catalyst for Suzuki coupling reactions previously reported [32, 34]. In the present case, during the synthesis event, various processes could occur simultaneously. The electroactive species (PdCl42− ions and GO flakes) are transported by the electrolyte convection and diffusion towards the rotating electrode; efficient transfer of electrons at the electrolyte/electrode interface for the electroreduction of species at very negative potentials; strong electroreduction of H3O+ ions to form hydrogen atoms and its combination to liberate molecular hydrogen (visible action); effective desorption of PdNps and rGO flakes from the electrode surface by the movement of reaction products towards the solution bulk, leaving free sites on the surface, and the non-aggregation control of particles in the dispersion, achieved by the presence of the stabilizing agent, which is in excess relative respect to the PdCl42− ion concentration.
A summary of the involved processes is shown in Scheme 1, where different species present in the aqueous electrolyte, such as PdCl42−, H3O+ ions, and GO flakes as well as the electroreduction products PdNps, H2, rGO flakes, and rGO–PdNps, respectively, are identified.

Graphical summary of the processes occurring during the two steps synthesis of rGO–AuPdNps hybrids in solution
As we mentioned, oxygen functional groups in the carbon structure of GO flakes reaching the electrode can be also electrochemically reduced. This process is indeed to occur at more negative potentials than –0.8 VSCE [35] for obtaining rGO–bidimensional flakes dispersion. The coincident approach scene of both species (PdCl42− ions and GO flakes) to the electrode/electrolyte interface can lead to the formation of supported rGO–PdNps in the solution, as shown by TEM images in Fig. 1. At the same time, H3O+ ions are consumed to form hydrogen adatoms, and the evolution of molecular hydrogen is abruptly produced, with a change in the pH value of the solution from 3.0 (initial state) to 3.5 (final state).

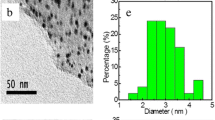
TEM (a), (b) and STEM (c) images with different magnifications of the PdNps supported on rGO flakes. (d) Size distribution histogram of Pd particles from STEM image, established with ImageJ software
rGO–PdNps formation was detected by the change in the solution coloration from pale yellow to dark brown after a few minutes after the beginning of the synthesis (see tube 1* in Fig. S1). As a result, PdNps are mostly incorporated into the structure of rGO flakes dispersed in solution. Both two-dimensional materials (GO and rGO) have low stability in a high ionic strength medium; however, their interaction with PVP molecules is very strong, such that its presence prevents the stacking of 2D particles [36]. Dispersion with individual structures remains stable for long periods, i.e., for several months no aggregation of the material was observed. Furthermore, final rGO flakes clearly act as support platforms for the Nps stabilized with PVP avoiding its polydispersion.
Morphology and distribution of PdNps on rGO–PdNps hybrids were studied by TEM and STEM images as it is shown in Fig. 1. Spherical particles of Pd are clearly incorporated into the laminar structure of rGO, aggregated in some sectors on the flake, not preferentially on the edges. Besides, many wrinkles from rGO flakes, i.e., the support platforms for Nps, are evidenced.
We can see that PdNps on rGO flakes are well spherically defined but they are rather polydisperse in size, as the histogram shows (Fig. 1d) an average diameter value of ~ 36 nm for PdNps in the rGO–Nps composites
Synthesis and characterization of the rGO–AuPdNps
After the electrochemical step where the rGO–PdNps hybrids dispersion was obtained, the controlled addition of HAuCl4 solution led to the formation of rGO–AuPdNps hybrids, through a galvanic replacement reaction. The rGO–PdNps dispersion was distributed in six test tubes (5.0 mL each), to which different aliquots of HAuCl4 solutions were added, and the chemical displacement reaction occurred on the PdNps surface where Au adatoms were deposited [37]. Table 1 shows the different amounts of 24 mM HAuCl4 precursor added to test tubes at the time of its addition. After 120 min of reaction, the change of color in the dispersions was notably appearing. Different colorations from each sample (Fig. S1), with violet (from gold nanoparticles) and brown (from partially reduced GO) colors superimposed in the dispersion, depending on the amount and size of AuNps formed in each tube. The presence of plasmonic particles (AuNps) on the rGO–PdNps hybrids and their morphology and size were analyzed by the optical properties of dispersions [28, 30].
Figure 2a shows the extinction spectra after 120 min of reaction between the AuCl4− ions and the rGO–PdNps dispersion, as indicated in Table 1 for test tubes shown in Fig. S1. The dependence of the absorption maximum at around 543 nm with the content of AuCl4− ions shows a wavelength shift for maxima surface plasmon absorption to higher values. There is also a shoulder at ~325 nm which is the characteristic of Pd and H2PdCl4, it has a poor definition due to the presence of PVP molecules and the rGO flakes [34, 38]. The appearance of the main feature is associated with the localized surface plasmon resonances (LSPR) sustained by the gold “decoration” formation during the galvanic exchange [39].
The evolution of successive extinction spectra during the galvanic replacement reaction is shown in Fig. 2b, recorded every 180 s after the HAuCl4 addition. Initially, the optical response is mainly due to the presence of PdNps on rGO, and the observed spectra show a fall of the extinction curve, for long wavelengths (Fig. 2a, red curve). During the first minutes, the galvanic replacement starts producing gold nuclei on the PdNps surface, the extinction in the range of 500–700 nm increases, and a broad plasmon band can be distinguished. During the reaction, for example in tube 6, a clear blue shifting of the LSPR maximum was evidenced, reaching values of ~543 nm at 120 min. The reason for this behavior could be that the morphology of the initial deposits is more like a thin shell rather than spherical nuclei.
In order to rationalize these experimental results, a series of finite difference time domain (FDTD) calculations have been carried out considering a single PdNp (50 nm diameter) with a single small Au bump, as a simplified model system. In Fig. 2c, it can be seen that there is an excellent correlation between the experimental and theoretical spectra. Even more, the molar fraction in each structure was calculated (Fig. 2d), leading to an approximated metal composition according to each optical response. This can be understood regarding the galvanic replacement reaction yield, as it was described by Xia and coworkers [40].
As mentioned above, obtaining the hybrid material with rGO flakes and AuPdNps does not require reducing agents and electrochemical control of the potential. In fact, the bimetallic structures are simply formed by a charge displacement that occurs on the PdNps surface. The charge transfer process involves the gold deposition through a galvanic exchange at specific sites, where the Pd atoms dissolution proceeds [27]. Thus, simultaneous nucleation of Au occurs spontaneously, as it is described by Eq. (1) to form the bimetallic AuPdNps as individual entities that remain deposited on the rGO flakes.
Features from GO and rGO in the absorption spectra, such as the electronic transitions π–π* of C = C bonds and n–π* of C = O bonds, at 230 and 300 nm [41], respectively, are not clearly defined in the spectra by the high absorbance at low wavelength. The distribution of AuPd bimetallic nanoparticles supported on rGO is surprisingly different from that observed for PdNps on rGO. Figure 3a, b shows a high density of bimetallic AuPdNps well distributed over the entire surface of the rGO flake, and some groups of spherical PdNps that have not yet been modified, are evidenced. The inhomogeneity in Nps size and distribution over the rGO flakes could be explained according to one of the models that Yang et. al. suggested for Au (Ag) Nps [42].

(a) Extinction spectra of rGO–PdNps dispersion after 120 min of reaction with different amounts of AuCl4− given in Table 1. (b) Evolution of extinction spectra from rGO–PdNps dispersion (sample of tube 6) during the spontaneous formation of AuNps for 120 min, by increasing the time of reaction. Successive spectra were recorded every 180 s. (c) Calculated far-field optical response of AuPd structures and, (d) the molar fraction of Au and Pd in each structure
TEM images show well-marked polyhedric shapes in Fig. 3c for bimetallic particles, where different shades of gray are given by the contrast of each material (Au and Pd) [35]. The size distribution histogram corresponding to the bimetallic nanoparticles from the sample in tube 6 is presented in Fig. 3d. In this case, the average diameter established was ~44 nm
The distribution of the bimetallic Nps on the carbon structure is markedly different. The population with smaller particles is higher in the bimetallic hybrid. The galvanic replacement reaction seems to produce an effect similar to an etching process of the Pd nanoparticle [43].
In order to establish the Au/Pd content ratio in the rGO–AuPdNps hybrids, EDS analysis on the sample of tube 6 (higher Au concentration), was performed. Figure 4a shows the EDS compositional spectrum of rGO–AuPdNps supported on a silicon wafer. It was recorded from a region with aggregation of bimetallic nanoparticles that allows the quantitative determination of Au and Pd elements on the sample, as is illustrated in Fig. 4b. Both metals are present in an Au/Pd ratio = 1.15 in these particular aggregates, indicating that the nanoparticles formed show a high percentage of Au covering the PdNps. This is important to indicate that different aggregates of rGO–bimetallic Nps hybrids evidence different chemical compositions, all of them with Au/Pd ratios a little higher than 1.0.
Crystallographic structures of different rGO–AuPdNps hybrids were determined from the XRD pattern of dried samples. Figure S2 shows the XRD pattern of two samples of rGO–AuPdNps obtained from different Au concentrations (tubes 4 and 6). Diffraction peaks at 2\(\theta\) = 38.4 and 44.7° are associated with (111) and (200) crystalline planes of the face-centered cubic (fcc) structure of Au, respectively [44]. Furthermore, the pattern presents two diffraction peaks at about 2θ = 39.3 and 46.8 associated with the (111) and (200), respectively, which are characteristic of the fcc structure of Pd [43]. A comparison between samples shows the increase of Au content on the PdNps surface given by the intensity of the Au (200) plane relative to that of (111) Pd crystalline planes.
The surface chemical composition of AuPdNps on the rGO support was analyzed by XPS. The spectra for the different hybrids are shown in Fig. S3. The binding energy (BE) for Pd0 3d5/2 normally appears at 334.9 eV, whereas PdOy species are usually found typically at 336.4 eV [45, 46]. Further Pd interactions, particularly the Pd–C appear at ~ 337.6 [47]. Moreover, the Au 4f7/2 peak normally appears at 83.8 eV while the two gold oxide states can be found at Au+ (BE of 85.6 eV) and Au3+ (BE of 87.3 eV) [48]. Thus, it was found that both rGO-PdNps and rGO-AuPdNps catalysts present peaks corresponding to Pd0 and Pd–C, due to the interaction of the Pd nanoparticles with the reduced graphene oxide support. Additionally, the rGO-PdNps catalyst shows peaks corresponding to Pd2+. On the other hand, the rGO-AuPdNPs catalyst presents Au 4f peaks that can be associated with the presence of Au0, Au+, and Au3+. This tendency of Au nanoparticles to oxidize may be due to a smaller nanoparticle size compared to Pd, which promotes the formation of oxides.
Spectroscopic characterization of the hybrids
In order to confirm the structure of reduced species of GO flakes after the electrochemical synthesis of PdNps, Raman spectra were recorded with laser radiation at 632.8 nm. In Fig. 5a, the Raman spectrum of rGO–PdNps dispersion displays bands at 1326.5 cm−1 and 1602.9 cm−1, from D and G typical signals of rGO, respectively. The G band corresponds to in-plane E2g mode from the vibration of sp2-bonded carbon atoms and the D band is associated with the structural defects on the carbon network. But also, the values of ID/IG intensity ratio from rGO–PdNps and rGO–AuPdNps are 1.17 and 1.3, respectively, indicating reduced structure, after the synthesis (the increase of ID/IG ratio is related to an average size of sp2 domains directly associated with D band intensity) [49,50,51]. Our data shows GO flakes were effectively reduced and possibly, their structural defects have increased due to the Nps inclusion in the sp2 carbon structure for both composites.
The spectroscopic characterization of the rGO-supported bimetallic (AuPd) nanostructures also reveals the presence of intensity-enhanced bands at 758.7 cm−1, 1425.6 cm−1, and 2929.2 cm−1 from CNC symmetric stretching and C–H vibrational stretching in the carbon chain of PVP molecules, respectively [52]. The observed SERS enhancements of the PVP molecules are attributed to the presence of the disseminated AuNps in the hybrid flake. When the laser illuminates the structure containing Au, induces a collective oscillation of free electrons (LSPR) [53], generating the enhancement of the electric field near the NP surface (hot spots). The Raman spectrum of the PVP molecules that are stabilizing the structures is then enhanced.
As it was mentioned, the calculated electric fields at 633 nm for PdNp and AuPdNp are compared in Fig. 5b, c. There is an increase in the maximum of the electric field (E/E0) nearby the surface in both structures: PdNp 3.5, AuPdNp 10 [39]. The last one exhibits better theoretical performance in SERS experiments, which has been proved experimentally (Fig. 5). Differences in the calculated Raman enhancements are proportional to [E/E0]4, in that case, the comparison between both samples gives values of approximately \(1.5 \times {10}^{2}\) and 104, where the structure containing Au deposits, will enhance the signal by almost two orders of magnitude more than the simple Pd structure.
Electrocatalytic activity of electrodes
In order to build robust platforms for checking the electrocatalytic activity, the rGO–Nps hybrid dispersions were deposited on a glassy carbon electrode (GC/rGO–Nps) without any binder polymer as Nafion [54]. It was performed by drop-casting of dispersion (50 μL) on the electrode and left to dry in the air. The presence of PVP molecules in excess is an advantage that favors the adhesion of particles on the electrode since they also act as a binder material.
Figure S4 shows the voltammetric experiments of Cu UPD performed in a deoxygenated CuSO4 electrolyte for the comparison of electrochemically active areas of each material deposited on the electrode. The results show the charge associated with the formation of a copper monolayer on the metallic nanoparticles in the hybrid structure (rGO-PdNps or rGO-AuPdNps) onto the modified electrodes, which was determined by the respective anodic dissolution peak (Table S1).
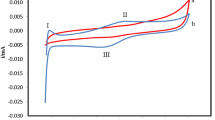
Figure 6a shows the cyclic voltammograms of rGO–PdNps and rGO–AuPdNps hybrids on glassy carbon in 0.5 M H2SO4 at 0.01 V s−1. Current densities were established taking to account the electroactive surface area of the electrodes, calculated by copper UPD measurements (Fig. S4) [55]. The voltammetric response of the carbon electrode in the absence of composite (GC, black dotted curve) shows a small current due to the hydrogen evolution reaction (HER) from –0.39 VSCE, without changes in successive cycles. The same trend is observed when we have a GC/rGO electrode without deposited nanoparticles. Onset potentials (OP) for HER on GC/rGO–PdNps and GC/rGO–AuPdNps electrodes were –0.31 VSCE and –0.24 VSCE, respectively (Table 2). OP values were calculated for the different electrodes when the current varies by 1 µA from baseline. We have observed that HER current was stable for GC/rGO–PdNps electrodes after the second voltammetric cycle, while a continuous increase in current density (read at –0.6 VSCE) was recorded during the first seven cycles for GC/rGO–AuPdNps electrodes (inset of Fig. 6).


(a) EDS compositional spectrum of a single rGO–AuPdNps flake from tube 6 deposited on a silicon wafer, and (b) table with compositional averaged content from different flakes from the sample in (a)

(a) Raman spectra with the ID/IG ratios of rGO–PdNps and rGO–AuPdNps dispersions (from tube 6). Black arrows indicate signals from vibrational modes of PVP molecules. Images of |E632.8 nm| calculated for a PdNp (b) and a PdNp with a single small Au–bump (c), showing the hot spots are located in the Au region

Cathodic potential scans (sixth cycle in the cyclic voltammetry) for GC, GC/rGO, GC/rGO–PdNps, and GC/rGO–AuPdNps electrodes in 0.5 M H2SO4 at 0.01 V s−1 scan rate. Current is normalized by the geometric area of the electrodes a) and real electrochemical area measured from Cu UPD b). Arrows indicate the onset potential values for HER. Inset: Evolution of the absolute current density at –0.6 VSCE in successive potentiodynamic cycles in the same range of potential with GC/rGO–AuPdNps electrode. c) Tafel plots of the overpotential vs. log j (current density) for the synthesized electrodes at the scan rate of 0.01 V s−1
The results indicate that the presence of AuNps in the rGO-PdAuNps hybrid generates changes in the resulting structures, which increases the catalytic activity with respect to rGO-Pd and rGO according to results presented in Fig. 6. This result may be due to the change of the Fermi level produced by the overlapping of the Pd atoms d band with the Au atoms d band [56, 57].
It can be seen that GC/rGO–AuPdNps electrodes have a catalytic activity greater than that of GC/rGO–PdNps (1.9 times greater current density at −0.6 V for the current density normalized by the electrochemically active area), indicating that there is an important activation process related to the decoration of PdNps with Au. This feature was evidenced in the lowest OP for HER with GC/rGO–AuPdNps (70 mV lower). Furthermore, an increase in the dj/dV slope was also distinguished (Table 2), with a value for rGO–AuPdNps 4.8 times greater than that for rGO–PdNps considering j normalized by the geometrical area. Moreover, when analyzing the dj/dV slopes ratio considering the j values normalized by the electrochemically active area, the effect is smaller with only a 1.4 times higher slope, indicating a clear effect of the change in the electrochemically active area on the electrochemical response. Besides that, both effects determine a noticeable catalytic activity produced by the presence of gold particles in the catalyst and the increase in the electroactive area that these particles generate. This change in the catalytic activity is evidenced by the current density at an overpotential of 200 mV where GC/rGO-AuPdNps is observed to have a jƞ200 one order of magnitude higher than GC and GC/rGO considering the geometrical area of the electrodes.
dj/dV is the ohmic resistance, OP is the onset potential of HER, 1/b is the reciprocal Tafel slope, and log j0 is the exchange current density.
Any changes in the HER mechanism occurring on these electrodes can be explained by the Tafel slope. Figure 6c shows the Tafel plots from the cyclic voltammograms of Fig. 6a for different electrodes. It can be seen that the Er decreases 40 mV when modifying the electrode of rGO–PdNps with AuNps. This result is in good agreement with the decrease in the value of OP presented in Table 2. The Tafel slope for the GC/rGO–PdNps electrode was –0.124 V dec−1, indicating that the transfer of one electron is the determinant step of the reaction rate (Volmer’s reaction). The same was found for the supporting materials, GC, and GC/rGO. In the case of GC/rGO–AuPdNps electrodes, the Tafel slope was –0.122 V dec−1 which possibly evidences the presence of coupled processes [58], consistent with the variation in current density observed during the stabilization period of the electrode. Values of exchange current density j0 from hybrids-electrodes were calculated (Table 2), and a noticeable increase of j0 of GC/rGO–AuPdNps electrodes was found, which explains the higher catalytic activity shown with the bimetallic composite. The increase of HER current density when the electrode contains the rGO-AuPdNps hybrid can be attributed not only to the surface roughness change but also to the electronic inductive effect given by Au on Pd with Fermi level shift [59]. In fact, both approximations can generate highly active sites for the electrocatalytic reaction [60].
Catalytic activity in comparison with literature
A comparison of our results with a variety of catalysts containing bidimensional nanomaterials for HER from literature using bidimensional nanomaterials [58, 61, 62] is presented in Table 3. Catalysts electrochemically synthesized such as GC/rGO–AuPdNps hybrids have a higher catalytic activity under conditions similar to other catalysts using bidimensional composites.
Although there is a wide variety of catalysts that take advantage of the enormous potential of the combination of metal-layered nanostructures, in Table 3, some of them were selected for comparison, where the mass activity is analyzed with respect to the amount of metal for the generation of hydrogen. It can be seen, in spite of some differences in the measurement conditions of the different materials that the hybrid nanomaterial catalysts presented in this work, particularly the rGO–AuPdNps, present mass activity values equal to or higher than those found in the literature, with the enormous advantage of the fast and reproducible synthesis method.
Conclusions
In summary, rGO–PdNps and rGO–AuPdNps hybrid nanomaterials were synthesized by a fast and reproducible method (combined electrochemical and galvanic replacement processes), without requiring heating, chemical reducing agents, subsequent purification, and washing. They can be directly deposited on surfaces for use on electrodes. This procedure allowed us to synthesize electrocatalysts for hydrogen generation in an acid medium with high efficiency, taking advantage of the particular characteristics of reduced graphene oxide. The synthesized electrodes show a greater efficiency toward the generation of hydrogen in an acid medium than other catalysts presented in literature containing two-dimensional nanostructures.
The as-obtained rGO–AuPdNps composites exhibit not only improved electrocatalytic ability for HER in acidic media but also have high sensitivity for adsorbed molecule detection. This is proved by the SERS spectra of the PVP molecules, which stabilize the composites in the aqueous dispersion.
It is important to note that the method presented here for the synthesis of catalysts is very simple, economic, and it can be extended to obtain different metal nanoparticles in solution. In this way, the proposed synthesis method allows the direct synthesis of catalytic ink to be placed on electrode supports without intermediate steps. It is a promissory type of composite that could be obtained in large-scale production and of course, be used in fuel cells for water splitting catalysis.
References
Anjana PM, Bindhu MR, Rakhi RB (2019) Green synthesized gold nanoparticle dispersed porous carbon composites for electrochemical energy storage. Mater Sci Energy Technol 2:389–395. https://doi.org/10.1016/j.mset.2019.03.006
Gualteros JAD, Garcia MAS, da Silva AGM, Rodrigues TS, Cândido EG, e Silva FA, Fonseca FC, Quiroz J, de Oliveira DC, de Torresi SIC, de Moura CVR, Camargo PHC, de Moura EM (2019) Synthesis of highly dispersed gold nanoparticles on Al2O3, SiO2, and TiO2 for the solvent-free oxidation of benzyl alcohol under low metal loadings. J Mater Sci 54:238–251. https://doi.org/10.1007/s10853-018-2827-x.
Vijilvani C, Bindhu MR, Frincy FC, AlSalhi MS, Sabitha S, Saravanakumar K, Devanesan S, Umadevi M, Aljaafreh MJ, Atif M (2020) Antimicrobial and catalytic activities of biosynthesized gold, silver and palladium nanoparticles from Solanum nigurum leaves. J Photochem Photobiol B Biol 202:111713. https://doi.org/10.1016/j.jphotobiol.2019.111713
Al-Thabaiti SA, Khan Z, Malik MA (2019) Bimetallic Ag-Ni nanoparticles as an effective catalyst for hydrogen generation from hydrolysis of sodium borohydride. Int J Hydrogen Energy 44:16452–16466. https://doi.org/10.1016/j.ijhydene.2019.04.240
Szustakiewicz P, Kolsut N, Leniart A, Lewandowski W (2019) Universal method for producing reduced graphene oxide/gold nanoparticles composites with controlled density of grafting and long-term stability. Nanomaterials 9:602. https://doi.org/10.3390/nano9040602
Liu M, Zhang R, Chen W (2014) Graphene-supported nanoelectrocatalysts for fuel cells: synthesis, properties, and applications. Chem Rev 114:5117–5160. https://doi.org/10.1021/cr400523y
Devadoss A, Sudhagar P, Das S, Lee SY, Terashima C, Nakata K, Fujishima A, Choi W, Kang YS, Paik U (2014) Synergistic metal-metal oxide nanoparticles supported electrocatalytic graphene for improved photoelectrochemical glucose oxidation. ACS Appl Mater Interfaces 6:4864–4871. https://doi.org/10.1021/AM4058925/SUPPL_FILE/AM4058925_SI_001.PDF
Cano M, Benito AM, Urriolabeitia EP, Arenal R, Maser WK (2013) Reduced graphene oxide: firm support for catalytically active palladium nanoparticles and game changer in selective hydrogenation reactions. Nanoscale 5:10189–10193. https://doi.org/10.1039/C3NR02822D
Zhou H, Chen L, Li S, Huang S, Sun Y, Chen Y, Wang Z, Liu W, Li X (2020) One-step electroreduction preparation of multilayered reduced graphene oxide/gold-palladium nanohybrid as a proficient electrocatalyst for development of sensitive hydrazine sensor. J Colloid Interface Sci 566:473–484. https://doi.org/10.1016/j.jcis.2020.01.105
Wang X, Gao H, Zhai C, He Z, Yuan C, Zhu M (2020) Newly found photoactivated Pt anchored on three-dimensional layered WS2/carbon cloth for highly efficient ethylene glycol electro-oxidation. Ind Eng Chem Res 59:19252–19259. https://doi.org/10.1021/acs.iecr.0c03436
Shi Y, Huang XK, Wang Y, Zhou Y, Yang DR, Bin Wang F, Gao W, Xia XH (2019) Electronic metal-support interaction to modulate MoS2-supported Pd nanoparticles for the degradation of organic dyes. ACS Appl Nano Mater 2:3385–3393. https://doi.org/10.1021/acsanm.9b00297.
Zhang XY, Li HP, Cui XL, Lin Y (2010) Graphene/TiO2 nanocomposites: synthesis, characterization and application in hydrogen evolution from water photocatalytic splitting. J Mater Chem 20:2801–2806. https://doi.org/10.1039/b917240h
Jung I, Dikin D, Park S, Cai W, Mielke SL, Ruoff RS (2008) Effect of water vapor on electrical properties of individual reduced graphene oxide sheets. J Phys Chem C 112:20264–20268. https://doi.org/10.1021/jp807525d
Guex LG, Sacchi B, Peuvot KF, Andersson RL, Pourrahimi AM, Ström V, Farris S, Olsson RT (2017) Experimental review: chemical reduction of graphene oxide (GO) to reduced graphene oxide (rGO) by aqueous chemistry. Nanoscale 9:9562–9571. https://doi.org/10.1039/c7nr02943h
Pei S, Cheng HM (2012) The reduction of graphene oxide. Carbon N Y 50:3210–3228. https://doi.org/10.1016/j.carbon.2011.11.010
Tarcan R, Todor-Boer O, Petrovai I, Leordean C, Astilean S, Botiz I (2020) Reduced graphene oxide today. J Mater Chem C 8:1198–1224. https://doi.org/10.1039/c9tc04916a
Abdin Z, Zafaranloo A, Rafiee A, Mérida W, Lipiński W, Khalilpour KR (2020) Hydrogen as an energy vector. Renew Sustain Energy Rev 120:109620. https://doi.org/10.1016/j.rser.2019.109620
Acar C, Bicer Y, Demir ME, Dincer I (2019) Transition to a new era with light-based hydrogen production for a carbon-free society: An overview. Int J Hydrogen Energy 44:25347–25364. https://doi.org/10.1016/j.ijhydene.2019.08.010
Yang T, Du M, Zhu H, Zhang M, Zou M (2015) Immobilization of Pt nanoparticles in carbon nanofibers: bifunctional catalyst for hydrogen evolution and electrochemical sensor. Electrochim Acta 167:48–54. https://doi.org/10.1016/j.electacta.2015.03.077
Shiraz HG, Shiraz MG (2017) Palladium nanoparticle and decorated carbon nanotube for electrochemical hydrogen storage. Int J Hydrogen Energy 42:11528–11533. https://doi.org/10.1016/j.ijhydene.2017.03.129
Darabdhara G, Amin MA, Mersal GAM, Ahmed EM, Das MR, Zakaria MB, Malgras V, Alshehri SM, Yamauchi Y, Szunerits S, Boukherroub R (2015) Reduced graphene oxide nanosheets decorated with Au, Pd and Au-Pd bimetallic nanoparticles as highly efficient catalysts for electrochemical hydrogen generation. J Mater Chem A 3:20254–20266. https://doi.org/10.1039/c5ta05730b
Franceschini EA, Lacconi GI (2018) Synthesis and performance of nickel/reduced graphene oxide hybrid for hydrogen evolution reaction. Electrocatalysis 9:47–58. https://doi.org/10.1007/s12678-017-0415-5
Franceschini EA, Lacconi GI, Corti HR (2017) Hydrogen evolution kinetics on Ni cathodes modified by spontaneous deposition of Ag or Cu. J Energy Chem 26:466–475. https://doi.org/10.1016/j.jechem.2016.10.009
Rakočević L, Štrbac S, Srejić I (2021) Hydrogen evolution on Au/GC and PdAu/GC nanostructures in acid solution: AFM, XPS, and electrochemical study. Int J Hydrogen Energy 46:9052–9063. https://doi.org/10.1016/J.IJHYDENE.2021.01.001
Ghasemi S, Hosseini SR, Nabipour S, Asen P (2015) Palladium nanoparticles supported on graphene as an efficient electrocatalyst for hydrogen evolution reaction. Int J Hydrogen Energy 40:16184–16191. https://doi.org/10.1016/J.IJHYDENE.2015.09.114
Rakočević L, Srejić I, Maksić A, Golubović J, Štrbac S (2021) Hydrogen evolution on reduced graphene oxide-supported PdAu nanoparticles. Catal 11:481. https://doi.org/10.3390/CATAL11040481.
Chen J, Lu ZH, Huang W, Kang Z, Chen X (2017) Galvanic replacement synthesis of NiPt/graphene as highly efficient catalysts for hydrogen release from hydrazine and hydrazine borane. J Alloys Compd 695:3036–3043. https://doi.org/10.1016/J.JALLCOM.2016.11.351
Makhafola MD, Modibane KD, Ramohlola KE, Maponya TC, Hato MJ, Makgopa K, Iwuoha EI (2021) Palladinized graphene oxide-MOF induced coupling of Volmer and Heyrovsky mechanisms, for the amplification of the electrocatalytic efficiency of hydrogen evolution reaction. Sci Rep 11.1:1–16. https://doi.org/10.1038/s41598-021-96536-9.
Johnson PB, Christy RW (1972) Optical constants of the noble metals. Phys Rev B 6:4370–4379. https://doi.org/10.1103/PhysRevB.6.4370
Palik ED (1985) Handbook of Optical Constants of Solids - 1st Edition, 1985th edn. Academic Press, New York
Weber JW, Calado VE, Van De Sanden MCM (2010) Optical constants of graphene measured by spectroscopic ellipsometry. Appl Phys Lett 97:091904. https://doi.org/10.1063/1.3475393
Uberman PM, Pérez LA, Martín SE, Lacconi GI (2014) Electrochemical synthesis of palladium nanoparticles in PVP solutions and their catalytic activity in Suzuki and Heck reactions in aqueous medium. RSC Adv 4:12330–12341. https://doi.org/10.1039/c3ra47854h
Bansala T, Mukhopadhyay S, Joshi M, An Doong R, Chaudhary M (2016) Synthesis and shielding properties of PVP-stabilized-AgNPs-based graphene nanohybrid in the Ku band. Synth Met 221:86–94. https://doi.org/10.1016/J.SYNTHMET.2016.07.034.
Uberman PM, Pérez LA, Lacconi GI, Martín SE (2012) PVP-stabilized palladium nanoparticles electrochemically obtained as effective catalysts in aqueous medium Suzuki-Miyaura reaction. J Mol Catal A Chem 363–364:245–253. https://doi.org/10.1016/j.molcata.2012.06.016
Marrani AG, Motta A, Schrebler R, Zanoni R, Dalchiele EA (2019) Insights from experiment and theory into the electrochemical reduction mechanism of graphene oxide. Electrochim Acta 304:231–238. https://doi.org/10.1016/j.electacta.2019.02.108
Swain AK, Bahadur D (2014) Enhanced stability of reduced graphene oxide colloid using cross-linking polymers. J Phys Chem C 118:9450–9457. https://doi.org/10.1021/jp500205n
Teng X, Wang Q, Liu P, Han W, Frenkel AI, Wen W, Marinkovic N, Hanson JC, Rodriguez JA (2008) Formation of Pd/Au nanostructures from Pd nanowires via galvanic replacement reaction. J Am Chem Soc 130:1093–1101. https://doi.org/10.1021/ja077303e
Shen H, Cheng BL, Lu GW, Guan DY, Chen ZH, Yang GZ (2005) Picosecond nonlinear optical responses of Au/PVP composite films. J Phys D Appl Phys 39:233. https://doi.org/10.1088/0022-3727/39/1/034
De Marchi S, Núñez-Sánchez S, Bodelón G, Pérez-Juste J, Pastoriza-Santos I (2020) Pd nanoparticles as a plasmonic material: synthesis, optical properties and applications. Nanoscale 12(46):23424–23443. https://doi.org/10.1039/D0NR06270G.
Chen J, Wiley B, McLellan J, Xiong Y, Li ZY, Xia Y (2005) Optical properties of Pd-Ag and Pt-Ag nanoboxes synthesized via galvanic replacement reactions. Nano Lett 5:2058–2062. https://doi.org/10.1021/nl051652u
Ding YH, Zhang P, Zhuo Q, Ren HM, Yang ZM, Jiang Y (2011) A green approach to the synthesis of reduced graphene oxide nanosheets under UVirradiation. Nanotechnology 22:215601. https://doi.org/10.1088/0957-4484/22/21/215601
Yang Y, Liu J, Fu ZW, Qin D (2014) Galvanic replacement-free deposition of Au on Ag for core-shell nanocubes with enhanced chemical stability and SERS activity. J Am Chem Soc 136:8153–8156. https://doi.org/10.1021/JA502472X/SUPPL_FILE/JA502472X_SI_001.PDF
Shon YS, Dawson GB, Porter M, Murray RW (2002) Monolayer-protected bimetal cluster synthesis by core metal galvanic exchange reaction. Langmuir 18:3880–3885. https://doi.org/10.1021/la025586c
Geraldes AN, Silva DF, Silva JCM, Souza RFB, Spinacé EV, Neto AO, Linardi M, Santos MC (2014) Glycerol electrooxidation in alkaline medium using Pd/C, Au/C and PdAu/C electrocatalysts prepared by electron beam irradiation. J Braz Chem Soc 25:831–840. https://doi.org/10.5935/0103-5053.20140044
Khawaji M, Chadwick D (2019) Selective catalytic oxidation over Au-Pd/titanate nanotubes and the influence of the catalyst preparation method on the activity. Catal Today 334:122–130. https://doi.org/10.1016/J.CATTOD.2018.11.080
Marx S, Baiker A (2009) Beneficial interaction of gold and palladium in bimetallic catalysts for the selective oxidation of benzyl alcohol. J Phys Chem C 113:6191–6201. https://doi.org/10.1021/JP808362M
Wagner C (1979) Handbook of x-ray photoelectron spectroscopy : a reference book of standard data for use in x-ray photoelectron spectroscopy. Physical Electronics Division Perkin-Elmer Corp Eden Prairie Minn
Sylvestre JP, Poulin S, Kabashin AV, Sacher E, Meunier M, Luong JHT (2004) Surface chemistry of gold nanoparticles produced by laser ablation in aqueous media. J Phys Chem B 108:16864–16869. https://doi.org/10.1021/JP047134
Tuinstra F (1970) KOENIG JL, Raman spectrum of graphite. J Chem Phys 53:1126–1130. https://doi.org/10.1063/1.1674108
Zhang Y, Guo L, Wei S, He Y, Xia H, Chen Q, Sun HB, Xiao FS (2010) Direct imprinting of microcircuits on graphene oxides film by femtosecond laser reduction. Nano Today 5:15–20. https://doi.org/10.1016/j.nantod.2009.12.009
Ferrari AC, Robertson J (2004) Raman spectroscopy of amorphous, nanostructured, diamond–like carbon, and nanodiamond. Philos Trans Royal Soc London. Series A: Math Phys Eng Sci 362(1824):2477–2512. https://doi.org/10.1098/rsta.2004.1452.
Mahmoud MA, Tabor CE, El-Sayed MA (2009) Surface-enhanced raman scattering enhancement by aggregated silver nanocube monolayers assembled by the Langmuir-Blodgett technique at different surface pressures. J Phys Chem C 113:5493–5501. https://doi.org/10.1021/jp900648r
Wustholz KL, Henry AI, McMahon JM, Freeman RG, Valley N, Piotti ME, Natan MJ, Schatz GC, Duyne RPV (2010) Structure-activity relationships in gold nanoparticle dimers and trimers for surface-enhanced raman spectroscopy. J Am Chem Soc 132:10903–10910. https://doi.org/10.1021/ja104174m
Viva FA, Bruno MM, Franceschini EA, Thomas YRJ, Ramos Sanchez G, Solorza-Feria O, Corti HR (2014) Mesoporous carbon as Pt support for PEM fuel cell. Int J Hydrogen Energy 39:8821–8826. https://doi.org/10.1016/j.ijhydene.2013.12.027.
Oviedo OA, Reinaudi L, Garcia S, Leiva EPM (2016). Underpotential deposition. https://doi.org/10.1007/978-3-319-24394-8
Zhang M, Hao Q, Yu Y (2013) Pd/Au(100) alloy for vinyl acetate synthesis: Effects of surface properties on reagents adsorption, Comput. Theor Chem 1019:33–38. https://doi.org/10.1016/J.COMPTC.2013.06.001
Quaino P, Santos E, Wolfschmidt H, Montero MA, Stimming U (2011) Theory meets experiment: electrocatalysis of hydrogen oxidation/evolution at Pd–Au nanostructures. Catal Today 177:55–63. https://doi.org/10.1016/J.CATTOD.2011.05.004
Hou D, Zhou W, Liu X, Zhou K, Xie J, Li G, Chen S (2015) Pt nanoparticles/MoS2 nanosheets/carbon fibers as efficient catalyst for the hydrogen evolution reaction. Electrochim Acta 166:26–31. https://doi.org/10.1016/j.electacta.2015.03.067
Pallassana V, Neurock M (2000) Electronic factors governing ethylene hydrogenation and dehydrogenation activity of pseudomorphic PdML/Re(0001), PdML/Ru(0001), Pd(111), and PdML/Au(111) surfaces. J Catal 191:301–317. https://doi.org/10.1006/jcat.1999.2724
Kelly CHW, Benedetti TM, Alinezhad A, Schuhmann W, Gooding JJ, Tilley RD (2018) Understanding the effect of Au in Au-Pd bimetallic nanocrystals on the electrocatalysis of the methanol oxidation reaction. J Phys Chem C 122:21718–21723. https://doi.org/10.1021/acs.jpcc.8b05407
Wang H, Lu Z, Kong D, Sun J, Hymel TM, Cui Y (2014) Electrochemical tuning of MoS2 nanoparticles on three-dimensional substrate for efficient hydrogen evolution. ACS Nano 8:4940–4947. https://doi.org/10.1021/nn500959v
Wang Y, Ma S, Li Q, Zhang Y, Wang X, Han X (2016) Hollow platinum nanospheres and nanotubes templated by shear flow-induced lipid vesicles and tubules and their applications on hydrogen evolution. ACS Sustain Chem Eng 4:3773–3779. https://doi.org/10.1021/acssuschemeng.6b00444
Acknowledgements
L.A.P. thanks CONICET for the postdoctoral fellowship. J.M.C. and F.F. thank EVC-CIN2020 students and SECyT-UNC doctoral fellowships, respectively. G.I.L. and E.A.F. are permanent research fellows of CONICET. The authors thank Dr. Ezequiel Leiva for very useful discussions on copper UPD measurements. This work was supported by projects from FONCyT PICT–2017-0250 (Argentina), CONICET PUE2017, and SECyT–UNC. The authors thank LAMARX, LANN-INFIQC laboratories for their assistance in XPS and SEM/EDX, Raman spectroscopy, and XRD measurements, respectively. G.I.L. thanks to the Alexander Von Humboldt Foundation for the equipment subsidy grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fioravanti, F., Pérez, L.A., Chierici, J.M. et al. Synthesis of rGO–Nps hybrids with electrocatalytic activity for hydrogen evolution reaction. J Solid State Electrochem 27, 61–74 (2023). https://doi.org/10.1007/s10008-022-05304-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-022-05304-w