
Abstract
N-doped graphene has been extensively explored because of their intriguing properties. However, most of the conventional heat-processed N-doped graphene (HNG) suffer from the poor hydrophilic property and low electric conductivity when using electrode materials. Herein, we present a facile solution-processed strategy to fabricate N-doped graphene through electrochemical exfoliation of graphite in inorganic electrolyte solution. The resulting electrochemically exfoliated N-doped graphene (ENG) has high level of nitrogen (7.9 at.%) and oxygen (16.5 at.%), moreover, excellent electric conductivity (19 s cm−1). As a binder-free electrode material for oxygen reduction reaction (ORR), ENG exhibits much better electroactivity than HNG and electrochemically exfoliated graphene (EG), moreover, much better methanol tolerance and long-term durability than that commercial Pt/C catalyst. The results provide new sights into scalable production of noble metal-free catalyst towards ORR.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the development of energy-related devices, fuel cell has been considered a promising approach in energy storage and conversion for its high efficiency and low emission [1]. Oxygen reduction reaction (ORR) is a critical process in fuel cell, which is normally catalyzed on cathode by Pt-based catalyst. However, the scarcity and sluggish kinetics of Pt-based material, together with the issues of instability and deactivation by CO and methanol crossover effect, have hampered the large-scale commercialization of fuel cell [2–4]. To overcome these obstacles, great efforts have been devoted in recent years to pursuing a broad range of alternative catalyst based on nonprecious metals or metal oxide decorated on carbon [5, 6], as well as metal-free doped carbon materials [7–10]. Metal or metal oxide nano-catalysts frequently suffer from the dissolution, sintering, and agglomeration [11, 12]. Currently, doping carbon materials with heteroatoms, such as N [13, 14], B [15], Fe [16], and Co [17], has been regarded as a promising strategy to prepare high efficent noble metal-free ORR electrocatalyst. Among these, nitrogen-doped carbon materials are particularly concerned owing to their excellent reliability and environmental friendliness.
Recently, graphene has been fascinated enormously as a new-generation carbon material because of its unique 2D planar structure, exceptional chemical, and physical properties. N-doped graphene (NG) is a very promising electrocatalyst for ORR because of their high catalytic activity [18–25]. NG is traditionally fabricated either by in situ carbonization of nitrogen-containing precursors [26, 27] or by post-treatment of the as-obtained carbon with a nitrogen-containing precursor such as NH3, CH3CN, and polypyrrole [28, 29]. However, for both in situ carbonization or post-treatment strategy, these heat-processable N-doped graphene materials (HNG) suffer from the poor hydrophilic property due to the low level of oxygen-containing, resulting in high interface energy between electrode materials with aqueous electrolyte. Consequently, the electrocatalytic activity of NG for ORR is still inferior to commercial Pt/C catalyst so far.
In recent years, electrochemically exfoliating graphite into graphene has attracted specific attention due to its easy, fast, and environmentally friendly nature. Graphite can be exfoliated in ionic liquids [30, 31] or aqueous acids (e.g., H2SO4 or H3PO4) [32, 33]. Generally, exfoliation in ionic liquids suffers from the low yield and, that in acidic electrolytes, can disrupt the structure of graphene due to the overoxidation induced by acids. Very recently, Müllen and Feng demonstrated that graphite can be electrochemically exfoliated in neutral inorganic salts, i.e., (NH4)2SO4, Na2SO4, and K2SO4 [34]. Inspired by this strategy, Zhao et al. developed a facile method to prepare NG through electrochemical exfoliation in NH4NO3 solution [35]. However, the N-doped level of this material is too low to catalysis ORR entirely. Herein, we intentionally designed a two-step electrochemical exfoliation strategy to prepare NG with abundant nitrogen and oxygen. In the first step of exfoliation, graphite paper is expanded in a weak exfoliation electrolyte, Na2SO4 aqueous solution. N-containing precursor, melamine-formaldehyde resin monomer (MFR), is subsequently permeated into the expanded interlayer of graphite sheets. After being calcined, nitrogen atom is doped into graphite paper. In the second step of exfoliation, the obtained N-doped graphite paper is further completely exfoliated in (NH4)2SO4 electrolyte. The resultant electrochemically exfoliated NG (ENG) presents outstanding electrical conductivity despite abundant oxygen species. As a binder-free electrochemical catalyst for ORR, ENG exhibits excellent electrocatalytic activity, remarkable long-term durability, and outstanding methanol tolerance.
Experimental section
Chemials
Graphite flake, graphite paper (99.95 %, 8000 mesh), Pt/C catalyst, melamine, and formaldehyde were purchased from Shanghai Aladdin Chemistry Co. Ltd. (China). All other chemicals were of analytical grade and used as received.
Preparation of electrochemically exfoliated graphene
One-step of electrochemical exfoliation strategy was employed to fabricated exfoliated graphene (EG). The exfoliation was performed in a two electrode system using stainless steel as the counter electrode and a graphite paper as the working electrode. Electrolyte solutions were prepared by dissolving (NH4)2SO4 in water (with a concentration of 0.1 M and pH 6.5–7.0). When a direct current (DC) voltage of +10 V was applied to the graphite electrode, the graphite flakes began to dissociate and disperse into the electrolyte solution. The electrochemical exfoliation apparatus can be found in Fig. S1. The voltage was kept constant for 4 h to finish the exfoliation process. Afterward, the exfoliated product was ultrasonically treated and collected by vacuum filtration and repeatedly washed with water to remove all residual salts. The resulting product was denoted as EG.
Preparation of MFR monomer
Melamine of 2.5 g and 6.6 mL of 37 wt% formaldehyde aqueous solution (with a molar ratio of 1:4.4) were added into 40 mL de-ionized water. After the solution was heat-treated at 70 °C for 10 min, the resultant transparent solution is MFR monomer.
Preparation of electrochemically exfoliated N-doped graphene (ENG)
Two-step of electrochemical exfoliation strategy was employed to fabricate ENG. The exfoliation process is similar to that of EG. Differently, the first step is the electrochemical expansion of graphite. The electrolyte in this step is sodium sulfate aqueous solution. Nitrogen-containing precursor, MFR monomer, was dissolved in the solution. A DC voltage of +10 V was applied to a graphite electrode. With graphite flakes expanding, MFR began to penetrate into the graphite sheets. The voltage was kept constant for 12 h, and the expanded graphite electrode was calcined in N2 atmosphere at 650 °C for 3 h to produce N-doped graphite paper. Afterward, the obtained N-doped graphite paper was used as the working electrode, and then was further electrochemically exfoliated in ammonium sulfate electrolyte solution under the equal condition with the first exfoliation step. The exfoliated product was ultrasonically treated and collected by vacuum filtration and repeatedly washed with water to remove any residual salts. The product prepared by this two steps of electrochemical exfoliation of graphite was denoted as ENG.
Preparation of heat-processed N-doped graphene (HNG)
Firstly, graphene oxide was prepared from nature graphite flakes by using an improved Hummers’ method [36]. Typically, graphite flakes (1.0 g) and KMnO4 (6.0 g) were added into mixture of 120 mL concentrated H2SO4 and 13.3 mL H3PO4, producing a slight exotherm to 35 °C. The mixture was then heated to 40~55 °C and stirred for 12 h. The reaction was cooled to room temperature and poured into ice water (150 mL) with 30 % H2O2 (10 mL). The mixture was sifted through a polyester fiber. The filtrate was centrifuged (4000 rpm for 4 h), and the supernatant was decanted away. The remaining solid material was then washed in succession with 200 mL of water, 200 mL of 30 % HCl, and 200 mL of ethanol. The eventual solution was centrifuged (4000 rpm for 4 h), and the supernatant was decanted away. The solid was vacuum-dried overnight at room temperature. Secondly, graphene oxide was hydrothermally treated with formaldehyde and melamine. The obtained composite hydrogel was freeze dried, and then calcined in N2 atmosphere at 750 °C for 6 h. The final product was denoted as HNG.
Construction of electrode
The procedures of glass carbon rotating disk electrode (RDE) (3 mm in diameter, from Autolab)—pretreatment and modification—are as follows: prior to use, the working electrode is polished mechanically with 0.5 μm diamond down to 0.05 μm alumina slurry to obtain a mirror-like surface and then washed with Mill-Q water and acetone and allowed to dry. Ten-milligram catalyst is dispersed in 5 mL water by sonication. The dispersion was dropped on the RDE and then allowed it to dry in ambient air to form a catalyst-modified RDE.
Electrochemical measurement
A conventional three-electrode cell was employed incorporating a working glass carbon RDE (Autolab) as working electrode, a Ag/AgCl electrode as reference electrode, and a platinum plate as counter electrode. All potentials were measured and reported vs the Ag/AgCl electrode and converted into vs the reversible hydrogen electrode (RHE). The experiments are carried out in O2-saturated 0.1 M KOH solution for the ORR. The potential range is cyclically scanned between 0.2 and +1.3 V vs RHE at a scanning rate of 2 mV s−1 at the ambient temperature after purging O2 or N2 for 15 min. RDE measurements are conducted at different rotating speeds from 400 to 2400 rpm by using an Autolab Model. The exact kinetic parameters including electron transfer number (n) and liminted kinetic current density (J k) were analyzed on the basis of Koutecky–Levich equations shown in equations.
\( \frac{1}{J}=\frac{1}{J_k}+\frac{1}{J_L}=\frac{1}{J_k}+\frac{1}{0.62nFA{D}_i^{2/3}{v}^{-1/6}{\omega}^{1/2}{C}_i} \)
where J is the measured current density, J k and J L are the kinetic and diffusion-limiting current densities, resectively, ω is the electrode rotation rate, n is the electron transfer number, F is the Faraday constant (F = 96,485 C mol−1), A is the area of electrode, C i is the saturated concentration of O2 (C i = 1.26 × 10−3 mol L−1) at room temperature, D i is the diffusion coefficient of O2 (D i = 1.9 × 10−5 cm2 s−1) at room temperature, v is the kinetic viscosity of the electrolyte (0.01 cm2 s−1) [37].
Characterization
The morphologies were examined with a JEM-2000 transmission electron microscope (TEM) with an acceleration voltage of 200 kV. All TEM samples were prepared by depositing a drop of diluted solution on a copper grid coated with carbon film. X-ray photoelectron spectroscopy (XPS) analysis was performed on a PHI Quantum 2000 Scanning ESCA Microprobe with a monochromatized microfocused Al X-ray source. The X-ray diffraction (XRD) patterns were measured with an X’Pert Philips Materials Research Diffractionmeter using Cu/Kα radiation.
Results and discussion
The preparation of ENG through electrochemical exfoliation route is schematically illustrated in Fig. 1. Firstly, graphite paper is electrochemically expanded in Na2SO4 electrolyte. The nitrogen-contained precursor, MFR monomer, is subsequently penetrated into the interlayer of expanded graphite. The MFR-decorated graphite paper is then dried in oven and calcined in N2 atmosphere under 650 °C for 3 h to perform N-doping. In comparison with the XRD pattern of graphite and N-doped graphite, a wider difraction peak centered at 26° is emerged, which is indicative of the expansion of graphite layers (Fig. S2). Elemental analysis by XPS reveals that the nitrogen content in N-doped graphite is ~4.3 % (Fig. S3). Secondly, the as-obtained N-doped graphite paper is electrochemically exfoliated in (NH4)2SO4 electrolyte. According to the exfoliation mechanism proposed by Müllen and Feng, the reduction of SO4 2− anions in Na2SO4 and self-oxidation of water produces gaseous species such as SO2, O2, and others [34]. These gaseous species will exert the force on the graphite layer, leading the expansion of the graphite. However, for (NH4)2SO4, much more gas species, i.e., NH3, can be produced, resulting in a stronger force to exfoliate N-doped graphite paper into thin layer graphene. The profile evolutions of graphite paper during the electrochemical exfoliation process are shown in Fig. S4.

Schematic illustration of the fabrication process of ENG. MFR represents melamine formaldehyde monomer here
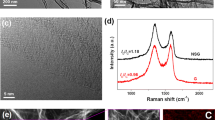
The morphology and structure of the as-prepared ENG were first examined under a scanning electron microscope (SEM) and transmission electron microscope (TEM). As shown in Fig. 2, SEM image displays a huge graphene sheet with a little crumple (Fig. 2a). The size of the graphene sheet even achieves to several micrometers. TEM images further confirm a layered graphene (Fig. 2b, c). Despite not single layer graphene, ENG still has thin-layer and integrate morphology. The corresponding selected area electron diffraction (SAED) pattern (Fig. 2d) demonstrates that ENG is crystalline [38].

a SEM, b, c TEM images of ENG, and d the corresponding selected area electronic diffraction
The structure of ENG was then characterized by XRD, N2 physical adsorption, and Raman spectroscopy. The XRD patterns are shown in Fig. 3a. For comparison, those of EG and HNG were recorded as well. It can be seen that all the XRD patterns exhibit a strong diffraction peak centered at 26.2°, which is assigned to the (002) diffraction peak [39]; moreover, their peak width is far broader than that of graphite (Fig. S2), which is indicative of a loosely layered structure of EG, ENG, and HNG [40]. As shown in Fig. S5, the adsorption-desorption isotherms of ENG and EG are the type II adsorption branch associated with a H3 hysteresis loop. Their BET surface areas are only 10.9 and 3.1 m2 g−1, respectively, suggesting that they are susceptible to be restacked in dry state. The Raman spectroscopy of ENG, EG, and HNG all emerges two characteristic bands at 1350 and 1581 cm−1 (Fig. 3b), corresponding to the D band and G band, respectively [41]. The G band is responsible for the in-plane vibration of sp2-hybridized C atoms, while D band reflects the defected graphene or the edge of perfect graphene. The value of ID/IG can be used to evaluate the structure integrity of graphene. As calculated, the ID/IG of EG, ENG, and HNG are 0.66, 0.89, and 1.05, respectively, demonstrating that electrochemically exfoliated strategy bears better structure than HNG chemical oxidation route. In addition, Raman spectroscopy of ENG displays another 2D band, while those of HNG are disappeared. The vanishing of the 2D band for the graphene sheet should result from the amorphization during the strongly oxidative steps [41].

a XRD pattern of EG, HNG, and ENG. b Raman spectroscopy of EG, ENG, and HNG
The chemical composition of ENG was further determined by XPS. As shown in Fig. 4, XPS survey (Fig. 4a) of ENG exhibits three pronounced peak centered at 285.7, 400.1, and 533.2 eV, corresponding to the characteristic peak of C, N, and O, respectively. The elemental analysis indicates that the contents of C, N, and O are 75.6, 7.9, and 16.5 %, respectively. In comparison with that of HNG and GO (Tab. S1), ENG has similarly high oxygen content with GO (~20 %), while approaching nitrogen content to HNG (~8 %). The high content of oxygen enables ENG with remarkable hydrophilic property. As shown in Fig. S6, a dispersion of ENG can maintain the original homogeneous state even after 10 days, however, that of HNG deposited completely in a day. The O1s XPS of ENG, HNG, and GO reveal that ENG has similar oxygen-related structure, but vary greatly from HNG (Fig. S7). The C1s XPS of ENG shows an absolutely dominated sp2-hybridized C peak (Fig. 4b), suggesting the good integrity of carbon skeleton. The N1s spectrum of ENG can be deconvoluted into three peaks located at the binding energy of 399.1, 400.2, and 402.1 eV, corresponding to the pyridinic, pyrrolic, and graphitic N, respectively (Fig. 4c) [21], which is similar to that of HNG (Fig. S8).

XPS survey (a), C1s XPS (b), N1sXPS (c) of ENG
The electric conductivity of ENG was finally tested via a four-probe method. As shown in Tab. S2, the electric conductivity of ENG achieves to 19.0 S cm−1, which is inferior to that of EG (33.4 S cm−1), due most likely to the doping of nitrogen [29]. However, the electric conductivity of ENG is much higher than those of HNG (1.75 S cm−1) and GO (5.32 × 10−4 S cm−1), despite large amount of oxygen. Furthermore, ENG can form paper-like material like GO through a facile filter treatment method, owing to the hydrogen bond interaction induced by the high content of oxygen species on its surface (Fig. S9), which sparks our interests that ENG may be an ideal bind-free electrode material. In this context, the electrochemical performance of ENG as a binder-free electrode material was investigated.
The electrocatalytic activity of ENG for ORR was firstly explored through cyclic voltammetry (CV) measurement with a conventional three-electrode mode. As shown in Fig. 5a, the CV curves of ENG in O2 and N2-saturated 0.1 M KOH aqueous solution at a scanning rate of 10 mV s−1 were compared. In the N2-saturated electrolyte, the CV of ENG shows a capacitive shape where there are no remarkable redox peaks. Despite low BET surface area in dry state, ENG still exhibits the normal specific mass capacity of 110 Fg−1, indicating that the area available for discharge in solution is much higher than that available for gas adsorption. In the O2-saturated electrolyte, there is a significant peak centered at 0.8 V in the O2-saturated electrolyte solution, which is the characteristic peak of ORR [42], suggesting that ENG has electrocatalytic activity towards ORR.

a Cyclic voltammograms of ENG at a scanning rate of 10 mV s−1 in N2 and O2-saturated 0.1 M KOH solution. b LSV of ENG in O2-saturated 0.1 M KOH solution with various rotation rates at a scanning rate of 2 mV s−1 (inset is the Koutecky-Levich plots of ENG derived from RDE LSVs at different electrode potentials)
To unravel the electrocatalytic mechanism of ENG for ORR, the kinetic behavior was investigated by RDE in O2-saturated 0.1 M KOH solution. Linear sweep voltammograms (LSVs) were recorded at the rotation speed ranging from 400 to 2400 rpm. The LSV scanning of ENG from both directions was firstly conducted, as shown in Fig. S10. It can be found that the optical scanning rate is 2 mVs−1. The LSV curves of all catalysts at the scanning rate of 2 mVs−1 were shown in Fig. 5b; all LSVs show typical higher current with increasing rotation speed, owing to the shortened diffusion distance at high speeds. Diffusion-limited current densities collected at different rotation speeds are used to determine the electron transfer number associated with ORR, which are calculated to be ~3.8 by the Koutecky-Levich equation (Fig. 5c). The electron transfer number is close to the theoretical four-electron transfer process, in which O2 is reduced into H2O by one step, suggesting that ENG is a remarkable ORR electrocatalyst. To maximize the ORR performance of ENG, the concentration of MFR monomer, the N-doping temperature, and the molar ratio of melamine to formaldehyde in MFR are systematically studied, as shown in Fig. S11. It is found that the optimal concentration of MFR, the N-doping temperature, and the molar ratio of melamine to formaldehyde in the precursor are 650 °C, 1:4.4, and 1.5 mol L−1, respectively.
The electrocatalytic performance of ENG was further compared with those of EG, HNG, and the commercial Pt/C catalysts (20 wt% on Vulcan XC72) (Fig. 6). As shown in Fig. 6a, the ORR onset potentials of ENG acquired from RDE linear sweep at 2400 rpm show that ENG had the onset potential of 0.92 V, which is significantly more positive than those of HNG (0.89 V) and EG (0.87 V). The kinetic current densities (J k, A/g) normalized by mass and their corresponding electron transfer number (n) are summarized in Fig. 6b. Remarkably, ENG exhibits a one-step, four-electron transfer pathway with a high kinetic current density of 2.6 A g−1. Although the electrocatalytic activity of ENG is worse than that of Pt/C catalyst (J k = 13.8 A g−1, n = 2.0), it is higher than those of EG (J k = 0.26 A g−1, n = 2.0) and HNG (J k = 1.80 A g−1, n = 2.8) significantly. The results confirm that ENG has much better electrocatalytic activity than EG and HNG, which may mainly attribute to the N-doping, hydrophilic surface, and good electric conductivity of ENG. Firstly, as reported, the lone electrons of the doped N can activate the π system of graphene, which make O2 molecular get reduced readily on the positively charged C atom neighboring N atom [43]; secondly, the hydrophilic surface induced by the high content of oxygen enhances the wettablity towards electrolyte, facilitating effective ionic transport. Finally, the excellent intrinsic electroconductivity and binder-free characteristic when using electrode material are in favor of the electron conduction. Additionally, the long-term durability of ENG was assessed through chronoamperometric measurements (Fig. 6c). ENG achieves much better durability with a high current retention (91.3 %) after 30,000 s of continuous operation compared to that of Pt/C (43.5 %). The cyclic stability of ENG and Pt/C was further evaluated by the current decay of the LSV curves at 2400 rpm (Fig. 6d). It can be seen that ENG has much better cyclic stability than Pt/C (Fig. S12). Considering the high activity and stability of ENG, it is expected that ENG is a promising electrocatalyst or support for ORR.

a RDE LSVs of ENG, HNG, EG, and Pt/C catalyst at the rotation speed of 2400 rpm and scanning rate of 2 mV s−1. b The calculated kinetic current density (J k) at 0.85 V, and average electron transfer number for EG, ENG, HNG, and Pt/C catalysts. c Chronoamperometric curves of ENG and Pt/C at the rotation speed of 2400 rpm. d LSV curves of ENG at 2400 rpm experienced 1000 and 5000 cycles. The loading of all the materials on the RDE is 0.20 mg cm−2
As an ORR electrocatalyst for fuel cells, a high catalytic selectivity for cathode reactions against fuel oxidation is of great significance, especially when using small molecule organic fuels, such as methanol and glucose in the anode, which could permeate through the polymer electrolyte membrane to the cathode, consequently seriously degrading the performance of the cell [44]. The methanol crossover effect was evaluated on ENG and commercial Pt/C catalysts (Fig. 7). An obvious cathodic current appeared when oxygen is purged into N2-saturated 0.1 M KOH aqueous solution at about 1000 s, indicating that ORR occurs on both ENG and Pt/C. After methanol is incorporated into the electrolyte, ENG still retains stable current response, whereas the current in the Pt/C system instantaneously jumped owing to the methanol oxidation reaction on Pt/C, revealing that ENG has better methanol tolerance than the vulnerable Pt/C. Consequently, ENG is an overall excellent electrocatalyst and has great potential to replace the classical Pt/C as a novel noble-metal-free catalyst.

Chronoamperometric responses at 0.75 V in N2-saturated 0.1 M KOH on ENG and Pt/C electrode followed by introduction of O2 and methanol (0.3 M)
Conclusions
In summary, we have developed a facile way to prepare NG. The resulting ENG exhibits greatly enhanced electrocatalytical activity for the cathodic ORR compared to the electrochemically EG and the conventional heat-processed NG in terms of electron transfer number, current density, and onset potential. The enhancement of electrocatalytic performance attributes to its excellent hydrophilic property, outstanding conductivity, and N-functionalization. ENG also shows superior long-term durability, cyclic stability, and methanol tolerance to the state-of-the-art Pt/C catalyst, suggesting that it is a promising electrocatalyst for fuel cell. Moreover, the proposed electrochemical exfoliation strategy is low-cost, eco-friendless, and suitable to scalable production, bringing hope to the industrialization of noble metal-free catalysts for ORR.
References
Choi HJ, Jung SM, Seo JM, Chang DW, Dai L, Baek JB (2012) Graphene for energy conversion and storage in fuel cells and supercapacitors. Nano Energy 1:534–551
Snyder J, Fujita T, Chen MW, Erlebacher J (2010) Oxygen reduction in nanoporous metal–ionic liquid composite electrocatalysts. Nat Mater 9:904–907
Zheng Y, Jiao Y, Jaroniec M, Jin YG, Qiao SZ (2012) Nanostructured metal-free electrochemical catalysts for highly efficient oxygen reduction. Small 8:3550–3566
Kareemulla D, Jayanti S (2009) Comprehensive one-dimensional, semi-analytical, mathematical model for liquid-feed polymer electrolyte membrane direct methanol fuel cells. J Power Sources 188:367–378
Oh HS, Kim H (2012) The role of transition metals in non-precious nitrogen-modified carbon-based electrocatalysts for oxygen reduction reaction. J Power Sources 212:220–225
Wu ZS, Yang S, Sun Y, Parvez K, Feng X, Müllen K (2012) 3D nitrogen-doped graphene aerogelsupported Fe3O4 nanoparticles as efficient electrocatalysts for the oxygen reduction reaction. J Am Chem Soc 134:9082–9085
Liang HW, Wei W, Wu ZS, Feng X, Müllen K (2013) Mesoporous metal-nitrogen-doped carbon electrocatalysts for highly efficient oxygen reduction reaction. J Am Chem Soc 135:16002–16005
Daems N, Sheng X, Vankelecom IF, Pescarmona PP (2014) Metal-free doped carbon materials as electrocatalysts for the oxygen reduction reaction. J Mater Chem A 2:4085–4110
Wei W, Liang H, Parvez K, Zhuang X, Feng X, Müllen K (2014) Nitrogen-doped carbon nanosheets with size-defined mesopores as highly efficient metal-free catalyst for the oxygen reduction reaction. Angew Chem 126:1596–1600
Niu WH, Li LG, Liu XJ, Wang N, Liu J, Zhou WJ, Tang ZH, Shao SW (2015) Mesoporous N-doped carbons prepared with thermally removable nanoparticle templates: an efficient electrocatalyst for oxygen reduction reaction. J Am Chem Soc 137:5555–5562
Chen Y, Li J, Mei T, Hu XG, Liu D, Wang J, Hao M, Li J, Wang J, Wang X (2014) Low-temperature and one-pot synthesis of sulfurized graphene nanosheets via in situ doping and their superior electrocatalytic activity for oxygen reduction reaction. J Mater Chem A 2:20714–20722
Jiang WJ, Hu JS, Zhang X, Jiang Y, Yu BB, Wei ZD, Wan LJ (2014) In situ nitrogen-doped nanoporous carbon nanocables as an efficient metal-free catalyst for oxygen reduction reaction. J Mater Chem A 2:10154–10160
Ai K, Liu Y, Ruan C, Lu L, Lu GM (2013) Sp2 C-dominant N-doped carbon sub-micrometer spheres with a tunable size: a versatile platform for highly efficient oxygen- reduction catalysts. Adv Mater 25:998–1003
Chen S, Bi J, Zhao Y, Yang L, Zhang C, Ma Y, Hu Z (2012) Nitrogen-doped carbon nanocages as efficient metal-free electrocatalysts for oxygen reduction reaction. Adv Mater 24:5593–5597
Choi CH, Chung MW, Kwon HC, Park SH, Woo SI (2013) B, N- and P, N-doped graphene as highly active catalysts for oxygen reduction reactions in acidic media. J Mater Chem A 1:3694–3699
Qiao X, Peng H, You C, Liu F, Zheng R, Xu D, Liao S (2015) Nitrogen, phosphorus and iron doped carbon nanospheres with high surface area and hierarchical porous structure for oxygen reduction. J Power Sources 288:253–260
Su Y, Jiang H, Zhu Y, Zou W, Yang X, Chen J, Li C (2014) Hierarchical porous iron and nitrogen Co-doped carbons as efficient oxygen reduction electrocatalysts in neutral media. J Power Sources 265:246–253
Luo Z, Lim S, Tian Z, Shang J, Lai L, MacDonald B, Lin J (2011) Pyridinic N doped graphene: synthesis, electronic structure, and electrocatalytic property. J Mater Chem 21:8038–8044
Zheng B, Wang J, Wang FB, Xia XH (2013) Synthesis of nitrogen doped graphene with high electrocatalytic activity toward oxygen reduction reaction. Electrochem Commun 28:24–26
Su Y, Zhang Y, Zhuang X, Li S, Wu D, Zhang F, Feng X (2013) Low-temperature synthesis of nitrogen/sulfur Co-doped three-dimensional graphene frameworks as efficient metal-free electrocatalyst for oxygen reduction reaction. Carbon 62:296–301
Kong XK, Chen CL, Chen QW (2014) Doped graphene for metal-free catalysis. Chem Soc Rev 43:2841–2857
Liang J, Jiao Y, Jaroniec M, Qiao SZ (2012) Sulfur and nitrogen dual-doped mesoporous graphene electrocatalyst for oxygen reduction with synergistically enhanced performance. Angew Chem Int Ed 51:11496–11500
Zhao Y, Yang L, Chen S, Wang X, Ma Y, Wu Q, Hu Z (2013) Can boron and nitrogen Co-doping improve oxygen reduction reaction cctivity of carbon nanotubes?. J Am Chem Soc 135:1201–1204
Yang Z, Nie H, Chen XA, Chen X, Huang S (2013) Recent progress in doped carbon nanomaterials as effective cathode catalysts for fuel cell oxygen reduction reaction. J Power Sources 236:238–249
Wang M, Wang J, Hou Y, Shi D, Wexler D, Poynton SD, Chen J (2015) N-doped crumpled graphene derived from vapor phase deposition of PPy on graphene aerogel as an efficient oxygen reduction reaction electrocatalyst. ACS appl Mater Interfaces 7:7066–7072
Ito Y, Christodoulou C, Nardi MV, Koch N, Sachdev H, Müllen K (2014) Chemical vapor deposition of Ndoped graphene and carbon films: the role of precursors and gas phase. ACS Nano 8:3337–3346
Lu YF, Lo ST, Lin JC, Zhang W, Lu JY, Liu FH, Li LJ (2013) Nitrogen-doped graphene sheets grown by chemical vapor deposition: synthesis and influence of nitrogen impurities on carrier transport. ACS Nano 7:6522–6532
Zhao Y, Hu CG, Hu Y, Cheng HH, Shi G, Qu LT (2012) A versatile, ultralight, nitrogen-doped graphene framework. Angew Chem Int Ed 51:11371–11375
Liu HT, Liu Y, Zhu DB (2011) Chemical doping of graphene. J Mater Chem 21:3335–3345
Liu N, Luo F, Wu HX, Liu YH, Zhang C, Chen J (2008) One-step ionic-liquid-assisted electrochemical synthesis of ionic-liquid-functionalized graphene sheets directly from graphite. Adv Funct Mater 18:1518–1525
Lu J, Yang JX, Wang JZ, Lim AL, Wang S, Loh KP (2009) One-Pot synthesis of fluorescent carbon nanoribbons, nanoparticles, and graphene by the exfoliation of graphite in ionic liquids. ACS Nano 3:2367–2375
Parvez K, Li RJ, Puniredd SR, Hernandez Y, Hinkel F, Wang SH, Feng XL, Müllen K (2013) Electrochemically exfoliated graphene as solution-processable, highly conductive electrodes for organic electronics. ACS Nano 7:3598–3606
Su CY, Lu AY, Xu YP, Chen FR, Khlobystov AN, Li LJ (2011) High-quality thin graphene films from fast electrochemical exfoliation. ACS Nano 3:2332–2339
Parvez K, Wu ZS, Li RJ, Liu XJ, Graf R, Feng XL, Müllen K (2014) Exfoliation of graphite into graphene in aqueous solutions of inorganic salts. J Am Chem Soc 136:6083–6091
Gondosiswanto R, Lu XY, Zhao C (2015) Preparation of metal-free nitrogen-doped graphene via direct electrochemical exfoliation of graphite in ammonium nitrate. Aust J Chem 68:830–835
Qin Y, Kong Y, Xu YY, Chu FQ, Tao YX, Li S (2012) In situ synthesis of highly loaded and ultrafine Pd nanoparticles-decorated graphene oxide for glucose biosensor application. J Mater Chem 22:24821–24826
Liang J, Zheng Y, Chen J, Liu J, Hulicova-Jurcakova D, Jaroniec M, Qiao SZ (2012) Facile oxygen reduction on a three-dimensionally ordered macroporous graphitic C3N4/carbon composite electrocatalyst. Angew Chem Int Ed 51:3892–3962
Zhang BB, Song JL, Yang GY, Han BX (2014) Large-scale production of high-quality graphene using glucose and ferric chloride. Chem Sci 5:4656–4660
Yang ZY, Zhao YF, Xiao QQ, Zhang YX, Jing L, Yan YM, Sun KN (2014) Controllable growth of CNTs on graphene as high-performance electrode material for supercapacitors. ACS Appl Mater Interfaces 6:8497–8504
Lee JH, Park NY, Kim BG, Jung DS, Im KY, Hur JH, Choi JW (2013) Restacking-inhibited 3D reduced graphene oxide for high performance supercapacitor electrodes. ACS Nano 7:9366–9374
Dresselhaus MS, Jorio A, Hofmann M, Dresselhaus G, Saito R (2010) Perspectives on carbon nanotubes and graphene raman spectroscopy. Nano Lett 10:751–758
Qin Y, Li J, Yuan J, Kong Y, Tao YX, Lin FR, Li S (2014) Hollow mesoporous carbon nitride nanosphere/three-dimensional graphene composite as high efficient electrocatalyst for oxygen reduction reaction. J Power Sources 272:696–702
Gong K, Du F, Xia Z, Durstock M, Dai L (2009) Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 323:760–764
Liu SH, Wu JR (2011) Nitrogen-doped ordered mesoporous carbons as electrocatalysts for methanoltolerant oxygen reduction in acid solution. Int J Hydrogen Energy 36:87–93
Acknowledgments
This work is supported by the National Natural Science Foundation of China (21476031, 21275023), the Advanced Catalysis and Green Manufacturing Collaborative Innovation Center of Changzhou University, the Priority Academic Program Development Project of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
ESM 1
(DOC 8055 kb)
Rights and permissions
About this article

Cite this article
Chao, L., Qin, Y., Liu, Y. et al. Electrochemically exfoliating graphite into N-doped graphene and its use as a high efficient electrocatalyst for oxygen reduction reaction. J Solid State Electrochem 21, 1287–1295 (2017). https://doi.org/10.1007/s10008-016-3480-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3480-4