
Abstract
The potential of new porous solid polymer electrolyte (SPE) for poly (vinylidene fluoride)–poly (methyl methacrylate) grafted natural rubber (PVDF-MG49) doped with LiCF3SO3 based on application in electrochemical device system has been investigated. The characteristics of the samples are analyzed and studied using electron impedance spectroscopy (EIS), X-ray diffraction (XRD), scanning electron microscopy (SEM), attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy, and linear sweep voltammetry (LSV). Ionic conductivity of 3.25 × 10−4 S cm−1 is achieved at room temperature, and the studies suggested that ion transport proceeds in these materials via a hopping mechanism similar to what is found in an ionic crystal. It is found that dielectric constant and dielectric loss increase with salt contents. A similar situation is also observed in electrical modulus. Analysis of XRD shows a decrease in crystallinity peaks of methyl methacrylate (MMA) in MG49 with the amount of added salt. The observations from SEM micrographs show porosity structure of polymer electrolyte. Based on the FTIR results, we are able to conjecture that interactions between the lithium ion and with the oxygen atoms from the MMA likely occur. Electrochemical studies show that polymer electrolyte has high electrochemical stability windows and is favorable for application in electrochemical devices.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ionically conducting materials such as solid polymer electrolyte play an important role in solid-state electrochemistry especially in energy storage (rechargeable battery and capacitor). The advancement and innovative inventions of electrochemical devices ranging from consumer electronic devices to hybrid electric vehicles (HEVs) are always critically depending on the development of battery technology. However, the battery performances always depends on the development of materials of the component parts in the battery [1]. One of the main components for flexible all solid-state lithium batteries is the solid polymer electrolyte (SPE). The SPE is widely regarded as an ideal solid electrolyte which provides freedom of cell assembly and safety, especially for larger size batteries [2]. However, the main drawback on practical use of solvent-free polymer electrolyte is low ionic conductivity at room temperature [3, 4].
To overcome this issue, a comprehensive study to improve the ionic conductivity has been done. Some of these approaches include polymer blending, modification of polymer structures, in situ thermal cross-linking, addition of inorganic oxide fillers (ZnO, Al2O3, TiO2, and NiO), gamma irradiation, UV irradiation, binary salts system, and use of plasticizers (EC and PC) [4–9]. Besides, Zhu et al. have tried to improve the ionic conductivity by proposing single-ion polymer electrolyte (lithium oxalate polyacrylic acid borate). Although the results showed low ionic conductivity, it achieved wide electrochemistry stability up to 7 V [10]. Among all these techniques, polymer blending is considered one of the most feasible techniques [2, 4, 6]. Aside from physical mixing, the blend of the two polymers or more is also achievable by means of simple photochemical reaction [11, 12]. The mixings of different polymers have created a new realm of technically important materials as their properties can be altered by varying the composition of the polymer blends. Although a large number of combinations of polymers are possible, there are relatively few leads to a totally miscible system [7].
The strong electron withdrawing and high dielectric constant of polyvinylidene fluoride (PVDF) has been one of the favorable choices for polymer electrolyte compositions. The semi-crystalline thermoplastic polymer has at least four possible polymorphs, namely, α-phase, β-phase, γ-phase, and δ-phase, due to its piezoelectric, ferroelectric, and pyroelectric properties with configuration structures. Depending on the annealing temperature (below 80–160 °C), poling voltage, and pressure, the most common and stable PVDF phases are α-phase and β-phase which are shown in the Fig. 1 [8, 13]. PVDF-based polymeric electrolyte has demonstrated sufficient conductivity value for commercial usage in secondary batteries. In the gel systems, the electrolytes based on PVDF are responsible for the ionic conduction and acted as the passive substrate/separator that retains the solution within the porous polymer framework [14]. Shen et al. have suggested that this porous PVDF also actively participates in the ion transport. In spite of the industrial importance and wide application of gelated PVDF, the ion conduction mechanism remains unsolved [15].

Schematic view of PVDF with alpha-phase (consists of a series of non-polar anti-parallel chains) and beta-phase phase (consists of a series of polar parallel chains)
Elastomer polymers, especially rubber material, are notable for its elasticity, soft elastomer nature at room temperature, and low glass temperature (T g). Polymer electrolytes based on rubber have been developed using modified natural rubber such as ENR50, LENR50, and MG30. In this research, 49 % poly (methyl methacrylate) grafted natural rubber (MG49) was chosen. MG49 consist of 49 % of poly (methyl methacrylate) (PMMA) grafted on rubber molecules. A typical polymerization reaction of MG49 is shown in Fig. 2. This modified grafted co-polymer of natural rubber latex and methyl methacrylate has gained attention due to its polar characteristics which serve as a conducting medium for transporting lithium ions and to achieve low T g for better chain mobility [4, 6, 7, 9, 16–20].

Typical polymerization reaction of rubber grafted with MMA monomer
In this work, we prepared and characterized the PVDF-MG49 polymer blend electrolytes with AC impedance spectroscopy, X-ray diffraction, Fourier transform infrared, scanning electron microscopy, and linear sweep voltammetry. Further studies were also done on ionic conductivity dependence upon temperature and electrical properties based on dielectric relaxation and modulus. We expect that the blending of soft elastomer MG49 which has good elasticity and adhesion properties with high porosity and mechanical strength PVDF will give a high ionic conductivity [17].
Materials and procedures
Materials
Forty-nine percent poly (methyl methacrylate) grafted natural rubber, MG49, used in this research was purchased from Green HPSP Malaysia (M w = 150,000). Poly (vinylidene fluoride), PVDF (M w = 180,000), lithium sheet (99.9 % trace metal), and lithium trifluoromethanesulfonate, LiCF3SO3 (156.01 g/mol), with 99.99 % purity were obtained from Sigma-Aldrich. Tetrahydrofuran (THF) was acquired from JT Baker.
Sample preparation
MG49 was cut into small pieces before subject to swell/dissolve in stoppered flasks containing THF for a night. The solution was then stirred with magnetic stirrer coated with Teflon for 24 h until complete dissolution of MG49 was obtained. In another new flask, PVDF was dissolved in THF and stirred for 24 h. These two solutions were then mixed together and kept under continuous stirring for 24 h. After a homogenous solution was obtained, the dissolved LiCF3SO3 salt was then added slowly to the mixed solution and continuous stirring was applied for another 24 h. The resulting homogenous polymer electrolyte solution was cast in a petri dish and allowed to evaporate slowly at room temperature in a fume hood until a film was obtained. The film was further dried in vacuum oven at 45 °C for 24 h to remove any residue solvent. The samples were then kept in desiccator before use for any characterizations. These steps were repeated for preparation PVDF-MG49 with 5–30 wt% of LiCF3SO3.
Sample characterizations
The ionic conductivity measurements were done at room temperature using alternate current (AC) impedance spectroscopy using high-frequency response analyzer (HFRA Solartron Schlumberger 1286) at a frequency of 0.1 to 1 MHz. The sample film was sandwiched between two stainless steel electrodes with a surface contact area of 2.01 cm2 and mounted it to the holder. Using the Z-View software, the bulk resistance R b of the sample film was determined and ionic conductivity was calculated based on equation σ = d/R b A, where d is the film thickness (cm) and A (cm2) is the effective contact area of electrolyte and the electrode. The data of complex impedance were used to study the dielectric constant and modulus. X-ray diffraction technique was conducted using Siemens model D5000 to determine the crystalline phase of the polymer electrolyte samples at diffraction angle of 2θ range from 2° to 60°. The X-ray radiation source used was CuKα with wavelength of 1.5418 Å. The morphological studies on the cross-section fractured surfaces of polymer electrolyte samples were done using SEM with ×5000 magnification at 15 kV electron beam. The attenuated total reflectance Fourier transform infrared (ATR-FTIR) analysis was performed using PerkinElmer Spectrum 2000 in the range of 4000 to 600 cm−1 with scanning resolution of 4 cm−1. The electrochemical stability window of the sample was evaluated using linear sweep voltammetry analysis in configuration of stainless steel/SPE/lithium. A voltage sweep was carried out on the material between 0 to 5 V at a constant rate of 10 mV s−1.
Results and discussions
Ionic conductivity studies
Figure 3a, b represents the complex ac impedance spectra of PVDF-MG49- LiCF3SO3 (5 to 30 wt%) electrolytes at room temperature. The electrolytes for 10 to 30 wt% of salt show nearly linear curves. The slanted spikes at high frequency region are attributed to the electrolyte resistance. This suggested that majority current carriers and conduction in the electrolyte are contributed by ions but not the electrons. On the other hand, Fig. 3c shows the variations of calculated ionic conductivity as a function to weight percentage of LiCF3SO3 salt contents for PVDF-MG49- LiCF3SO3 electrolytes. From the graph, it is observed that ionic conductivity increased with lithium salt contents. The highest ionic conductivity of 3.25 × 10−4 S cm−1 is achieved at 30 wt% of LiCF3SO3. The increase in the ionic conductivity is due to increases in charge carriers in the matrix electrolyte system. This can be shown by the equation σ = ce(u + + u −) = cΛ where σ is the conductivity of electrolyte, Λ is molar conductivity, c is salt concentration, e is charge of an electron, and u + and u − represent the ion mobility. As number of charges increase, the ionic conductivity also increases [4, 6]. However, further addition of lithium salt above 30 wt% causes the sample to be unstable (not free-standing). This might be due to the large anions from the lithium salt acts as a plasticizer by interrupting the ordered arrangement of the polymeric backbones and thus reduces the crystallinity in the electrolytes. The reduction of crystallinity is shown in analysis of X-ray diffraction (XRD). This means that more free spaces and voids are created for ion migrations. It is worth to mention on a steep increase in ionic conductivity at low concentration of salt loading before it slowly increases at higher salt contents as shown in Fig. 3c. In this case, at low concentration of salt, the ionic conductivity is strongly affected by the number of charge carriers as mobility of ions is relatively uninfluenced. Contrary, at higher concentration of salt, ionic conductivity is more strongly dependent on the ionic conduction pathways and ion mobility [21, 22].

AC impedance spectra for SPE PVDF-MG49- LiCF3SO3 with a 5 wt% and b 10–30 wt% lithium salt contents; c variation of ionic conductivity as a function of weight percentage of LiCF3SO3 salt in PVDF-MG49- LiCF3SO3 complexes
Temperature dependence–ionic conductivity studies
Figure 4 depicts the dependence conductivity on the temperature analysis by the Arrhenius plot for PVDF-MG49-LiCF3SO3 (30 wt%) to study the transportation property of the polymer electrolyte. At the fixed concentration of salt, the temperature dependence–ionic conductivity plot displays a linear relationship with the regression value obtained is close to unity (with regression coefficient of 0.9946). This suggests that the semi-crystalline polymer electrolyte followed the Arrhenius rules in which cation transport property is ionic hopping mechanism, similar to that in an ionic crystal where ions jump into the neighboring vacant site with well-defined pathways. The estimated activation energy (E a) is valued at 0.14 eV. Contrary to amorphous polymer electrolytes, the ion transportation is much influenced by the dynamic formation of suitable coordination sites between which ions can migrate [23].

Arrhenius ionic conductivity plot of PVDF-MG49-LiCF3SO4 (30 wt%)
This phenomena can be explained by ion hopping mechanism happens between coordinating sites and local structural relaxation. As the temperature increased, the vibrational modes of polymer segments increased and gained sufficient energy to push against the hydrostatic pressure, which is imposed by its surrounding atoms and turns to the formation of voids [24]. At the same time, the polymers will experience expansion effect, resulting in more local vacant spaces and creates more free volumes for segmental motions. Subsequently, this weakens the interactions between the polar group of the polymer backbones and Li+ and thus promotes the decoupling process of the charge carriers from the segmental motion of the polymer matrix. This facilitates the migration of ions and diminishes the ion cloud effect between the electrodes and electrolyte interface. These two entities (vacant sites and polymer segmental motions) then permit free charges to hop from one site to another thus increase conductivity. Therefore, the neighboring ions from adjacent sites tend to occupy these vacant sites and coordinate with the polymer chains again. Eventually, the ionic hopping mechanism is generated. It is worth noting that no abrupt change in the value of conductivity with respect of temperature was observed, inferring no phase transitions within the temperature regime for all the complexes [25].
Overall, the mechanism of PVDF-MG49-LiCF3SO3 complex in lithium ion polymer battery can be illustrated as in Fig. 5.

Illustration of typical working principle of lithium ion polymer battery and mechanism of SPE for PVDF-MG49-LiCF3SO3
Dielectric relaxation studies
Dielectric relaxation studies are vital tool to gain valuable information about the conduction processes since they can be used to understand the origin of the dielectric losses, electrical and dipolar relaxation time, and its activation energy in the polymer electrolyte system. It is a ratio of the permittivity of a substance to the permittivity of free space [26–29]. Generally, the dielectric constant is a measure of how much the charge stored. The cause of the dielectric constant is due to electrode polarization effects, space charge effects (normally observed in ionic glasses), and also water trace that may be retained by the films. The dielectric permittivity is determined from the measured values of the impedance using the following relation [26, 27, 30–32]:
where Z’ is the real part of complex impedance, Z” is the imaginary part of complex impedance, ω is the angular frequency (2πf), C o is vacuum capacitance (ε o × A/d), and ε’ and ε” are the real and imaginary components of the dielectric permittivity, respectively. ε o is the permittivity of free space (8.85 × 10−12 F/m), A is area, and d is the thickness of the electrolyte.
Figure 6 shows the frequency dependent plot of ε’ and ε” (also known as dielectric loss) parts of dielectric permittivity curves. From the graph, it is observed that both ε’ and ε” increase toward low frequency and gradually slowly approaching zero at high frequency. As the amount of salts increases, the dielectric constant increases. The highest value for both ε’ and ε” is observed at 30 wt% salt. The increase of dielectric constant with respect to salt content is due to the enhanced of charge carrier density in the space charge accumulation region, which caused the equivalent capacitance enhancement [29]. This means that PVDF-MG49- LiCF3SO3 (30 wt%) has the maximum number of charge stored. Since the graph does not show any relaxation peaks, this indicates that the increase in conductivity value is due to increase in free charge carriers [33]. The same trend for dielectric loss (ε’) is observed as for dielectric constant. No appreciable peaks were observed.

Variations of real (ε’) part and imaginary (ε”) of dielectric constant with frequency for SPE PVDF-MG49- LiCF3SO3 at different salt contents
This situation can be explained according to the behavior of the dipole movements with the dielectric permittivity related to the free dipoles oscillating in the presence of an alternating electric field. At the very low frequencies (f < 1/τ, where τ is the relaxation time), dipoles follow the electric field. As the frequency increases, dipoles begin to lag behind the field and slightly decrease. When the frequency reaches the characteristic frequency (f = 1/τ), the dielectric constant drops (relaxation process). At the very high frequencies (f > 1/τ), dipoles can no longer follow the field and ε’ ≈ ε’∞. Higher values of dielectric constant at the low frequencies can be explained as the formation and accumulation of the charges at the grain boundaries and at the interfaces between the electrolyte and the electrode (known as space charge polarization). This is well known as the non-Debye type of behavior, where the ion diffusion explains the space charge regions with respect to the frequency.
Dielectric modulus studies
Similarly, the real and imaginary parts of modulus functions are expressed by the following [16–27, 30, 31]:
In order to reveal the relaxation peak in ε” plot, the effect of the electrode polarization must be excluded. This can be achieved by studying the dielectric modulus plots since the dielectric modulus (M*) corresponds to the relaxation on the electric field in the material when the electric displacement remains constant [26]. In this case, the appearance of the peak in the imaginary part of dielectric modules can be related to transition ionic dynamics and the conductivity relaxation of the mobile ions [31]. Another purpose is to support the electrical conductivity data. The advantages of modulus studies were to recognize the conduction process and the effect on the frequency [27]. Figure 7 shows the frequency dependence of the real part (M’) and imaginary part (M”) of dielectric modulus. With increasing lithium salt content, the value of relaxation peaks shifted to the higher frequency. The distribution of the relaxation peaks is due to the distribution of relaxation time of the free charge. The existence of the relaxation peak curves at the higher frequencies in the real part modulus indicates that the polymer electrolyte films are ionic conductor [28, 29, 32]. The relaxation peak does not exist in the imaginary modulus graph which is because the residual water does not contribute toward conductivity enhancement [27].

Variations of the M’ part and M” part with frequency for SPE PVDF-MG49- LiCF3SO3 at different salt contents
It is observed that M’ and M” decrease when approaching low frequencies due to the electrode polarization phenomenon. This contribution of electrode polarization can be ignored at lower frequencies of M’ and M”. Furthermore, the tail was observed in the lower part of the frequency which indicates that the samples are capacitive in nature for M’ and the distribution of relaxation time of the ionic conductor for M”. The existence of the long tail can be considered as the result of the large capacitance associated with the electrode [27].
Analysis XRD
X-ray diffraction measurements are conducted to examine the nature crystallinity and the occurrence of complexation in polymer electrolyte in the presence of various salt concentrations. The crystalline phase is depicted in Fig. 8, showing that crystalline peaks decreased as salt dopant increased. The semi-crystalline nature of PVDF was confirmed by the appearance of the peaks at 2θ = 18.3°, 20.1°, and 26.5° while MG49 crystalline peak was observed at 2θ = 29.5° [34, 35]. The sharp peak at 20.09° is attributed to the presence of β-polymorph of PVDF. From the diffractogram, significant change in peak intensity was observed at 2θ = 29.5°. As the salt concentration increased, the intensity peak decreased [17, 18]. This indicates that the polymer electrolytes undergo structural reorganization leading to decrease in crystallinity and complexation occurring at oxygen-based MMA group which comprises of oxygen group (further supported by FTIR results) [34]. Moreover, the salt is well-solvated in the polymer matrix as no crystalline peaks of lithium salt are observed [4, 6, 9, 34, 36, 37].

XRD patterns of a PVDF, b MG49, c PVDF-MG49, d SPE-10 wt% of LiCF3SO3, e SPE-20 wt% of LiCF3SO3, f SPE-30 wt% of LiCF3SO3, and g pure salt LiCF3SO3
The relationship between degree of amorphousity and ionic conductivity can be related to the greater ionic diffusivity and mobility of ions in the amorphous phase because of low energy barriers. In amorphous phase, polymer backbones are more flexible and have higher segmental chains motion. This segmental motion in amorphous phase promotes ion motion by making and breaking the coordination sphere of the solvated ion and providing space (free volume) into which the ion may diffuse under the influence of the electrical field [4, 6, 9].
Morphological studies
The SEM micrographs in Fig. 9 are taken at the cross-sectional areas for SPEs with different amounts of LiCF3SO3 salts at magnification of ×5000. In Fig. 9a, b, the smooth surfaces of MG49 and PVDF are shown, respectively. After blending together (see Fig. 9c), the surface morphology drastically changed and became rougher. It is observed that most surfaces are surrounded by small regular round shape PVDF and have phase separation which suggested that the polymer blend is compatible but not miscible. The immiscible blend is proven by XRD patterns in which the PVDF-MG49 blend (in Fig. 8c) shows distinguishable peaks of PVDF and MG49. It is worth to note that the crystalline peak of MMA at 2θ = 29.5° has lower intensity when compare to crystalline peak in pure MG49. The above results suggested that PVDF is not miscible with MG49. This can be explained by law of thermodynamics through Gibbs free energy and also entropy in which the relationship governing mixtures of dissimilar components can be represented by the following equation [6]:

SEM micrographs at cross-sectional morphology of a MG49, b PVDF, and SPEs PVDF-MG49 doped c 0 wt%, d 10 wt%, e 20 wt%, and f 30 wt% of LiCF3SO3 salt
where ΔG m is free energy of mixing, ΔH m is the enthalpy of mixing (heat of mixing), and ΔS m the entropy of mixing. A blend of two components is classified as miscible, thermodynamically, if the overall change of Gibbs free energy of the mixing is negative [4].
The cross-sectional views of SPEs doped with different salt concentration are shown in Fig. 9d–f. After introducing the lithium salt, interconnected pores were observed. The presence of micro-pores formed through the complex process of interaction between solvent, lithium salt, and the polymers during evaporation. Furthermore, the amount of pores and its sizes are influenced by the difference in driving force of phase separation and kinetically controlled by the relative rate of evaporation of the compounds [4, 6, 9]. Nevertheless, the formation of the pores in the polymer–salt matrix will aid the mobility of ions by providing and creating more pathways for ions transportation [34, 38]. When the concentration of salts increases, the surface morphology becomes smoother [18]. This implied that salt helps promote compatibility of PVDF-MG49 in the complex matrix. As mentioned before, the salts disrupt the chain organizations in the polymer and subsequently decrease the crystalline phase. The increase in amorphousity which has a higher state of entropy will help in polymers mixing. One of the reasons polymers is mixable is because they are more disordered mixed together than when they are separate. This is proven in XRD patterns in Fig. 8c where the mixed polymers have lower intensity in crystallinity peaks compared to their respective individual single polymer.
Analysis ATR-FTIR
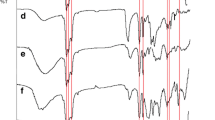
Complex formation and local structural changes in the polymer electrolyte matrices were determined from analysis of ATR-FTIR. Figures 10, 11, and 12 elucidate the FTIR spectra of SPEs at various concentrations of lithium salt. In Fig. 10, the detected peaks are CH2 rocking mode, CH2 twisting mode, symmetric stretching CF2, asymmetric stretching C–O–C group, and symmetric stretching CF2, respectively, at bands 840, 976, 1065, 1146, and 1181 cm−1 [17, 18, 29, 37, 39]. Others peaks that were observed are amorphous phase of PVDF at 872 cm−1 and free SO3 − of lithium salt at 1032 cm−1 [29, 30]. When the salt concentration increased, the intensity peaks for amorphous phases, SO3 − anion, and v s -CF2 of PVDF were increased. Besides, intensity peak of v as C–O–C group was found to be decreased and shifted to 1152 cm−1. This implied complex reaction has occurred at oxygen from ester group [32].

FTIR spectra (1200–800 cm−1) of PVDF-MG49-LiCF3SO3 (0, 10, 20, and 30 wt%)

FTIR spectra (1800–1200 cm−1) of PVDF-MG49-LiCF3SO3 (0, 10, 20, and 30 wt%)

FTIR spectra (3800–2700 cm−1) of PVDF-MG49-LiCF3SO3 (0, 10, 20, and 30 wt%)
In Fig. 11, a drastic drop in intensity was observed for carbonyl group, C=O at band 1728 cm−1 while the v as C–O group peak at 1280 cm−1 almost disappeared when concentration of salt increased [35, 37, 39]. In addition, a new peak evolves at the lower frequency (1645 cm−1) and its intensity increases with salt concentration. According to Kumutha et al., this band corresponds to Li+→O=C interaction [17]. On other hand, it is found that the deformation mode of CH2 and O–CH3 at 1435 cm−1 and 1448 cm−1 was shifted to lower frequency at 1425 cm−1 and 1454 cm−1 respectively [17, 18, 34, 39]. The changes in intensity, emerging of new peak, and peak shifting, especially at C–O–C, C=O, O–CH3, and C–O groups indicated that lithium ion salt prefers to undergo a complex interaction with the oxygen group.
Figure 12 shows that a broad peak corresponding to hydroxyl group, v(OH), was observed and increases with salt concentration which is due to the hydroscopic nature of lithium salt at around 3400 cm−1 [35]. The infrared bands at 2986, 2972, 2955, and 2853 cm−1 are assigned respectively to symmetric stretching CH2, CH3, asymmetric stretching CH, and symmetric stretching CH [35, 37, 39, 40]. Overall, the intensity of peaks in region 2853–2986 cm−1 decreased when lithium salt was introduced. Notably, the v s CH2 at band 2986 cm−1 was shifted to 3027 cm−1 and became v asCH2.
LSV study
For application in electrochemical devices, it is important for polymer electrolyte to have high electrochemical stability. This is an essential parameter because the instability of electrolyte will cause irreversible reactions and capacity fading [41]. Figure 13 shows the linear sweep voltammetry (LSV) curve for PVDF-MG49-LiCF3SO3 (30 wt%). The anodic decomposition limit of the polymer electrolyte is considered as the voltage at which the current flows through the cells. As can be seen from the plot of current versus voltage, the electrolyte shows anodic stability of 3.0-V windows. The high electrochemical stability window is a valuable property for the fabrication of stable and durable electrochemical devices.

Linear sweep voltammogram of PVDF-MG49-LiCF3SO3 (optimum, 30 wt%) based polymer electrolyte
Conclusion
Solid polymer electrolyte PVDF-MG49-LiCF3SO3 has been successfully prepared via solution casting technique. The results showed that ionic conductivity increases with salt concentration and the highest value of ionic conductivity was obtained at 3.25 × 10−4 S cm−1 for 30 wt% of salt. Further studies indicated that ion transport of polymer electrolyte is involved in ion hopping mechanism. Dielectric study revealed that dielectric constant increases with salt concentration toward the end of low frequency. Dielectric modulus also increases, but toward the high frequencies. XRD results showed that crystalline decreases with concentration of salt. The increase in amorphousity have enhanced the ionic conductivity and improved the overall morphological structures of the SPE due to increase compatibility of salt with polymer blends. Evidence of the complex interaction occurred between the oxygen group of MG49 and lithium ion was observed in FTIR spectra. The electrochemical stability window shows that the SPE is suitable to apply in electrochemical devices.
References
Arbizzani C, Gabrielli G, Mastragostino M (2011) Thermal stability and flammability of electrolytes for lithium-on batteries. J Power Sources 196(10):4801–4805
Fergus JW (2010) Recent developments in cathode materials for lithium ion batteries. J Power Sources 195(4):939–954
Bruce PG (1995) Solid state electrochemistry. Cambridge University Press, Great Britain
Lee TK, Ahmad A, Dahlan HM, Rahman MYA (2012) LiClO4 salt concentration effect on the properties of PVC-modified low molecular weight LENR50-based solid polymer electrolyte. J Appl Polym Sci 124:2227–2233
Tripathi BP, Shahi VK (2011) Organic–inorganic nanocomposite polymer electrolyte membranes for fuel cell applications. Prog Polym Sci 36(7):945–979
Lee TK, Afiqah S, Ahmad A, Dahlan HM, Rahman MYA (2012) Temperature dependence of the conductivity of plasticized poly (vinyl chloride)-low molecular weight liquid 50% epoxidized natural rubber solid polymer electrolyte. J Solid State Electrochem 16:2251–2260
Perera MCS, Ishiaku US, Ishak ZAM (2001) Characterisation of PVC/NBR and PVC/ENR50 binary blends and PVC/ENR50/NBR ternary blends by DMA and solid state NMR. Eur Polym J 37:161–178
Mago G, Kalyon DM, Fisher FT (2008) Membranes of polyvinylidene fluoride and PVDF nanocomposites with carbon nanotubes via immersion precipitation. Journal of Nanomaterials pp 1–8
Rahman MYA, Ahmad A, Lee TK, Farina Y, Dahlan HM (2011) Effect of ethylene carbonate (EC) plasticizer on poly (vinyl chloride)-liquid 50% epoxidised natural rubber (LENR50) based polymer electrolyte. Mate Sci Appl 2:817–825
Zhu YSX, Gao W, Wang XJ, Hou YY, Liu LL, Wu YP (2012) A single-ion polymer electrolyte based on boronate for lithium ion batteries. Electrochem Commun 22:29–32
Gianmarco G, Federico B, Filippo N, Claudia D, Dominique R, Marinella L, Roberta B, Stefano T (2015) Multifunctional luminescent down-shifting fluoropolymer coatings: A straightforward strategy to improve the UV-light harvesting ability and long-term outdoor stability of organic dye-sensitized solar cells. Adv Energy Mater 5:1401312
Adriano S, Federico B, Stefano DLP, Micaela C, Stefano B, Roberto B, Candido FP (2015) Electrodes/electrolyte interfaces in the presence of a surface-modified photopolymer electrolyte: Application in Dye-sensitized solar cells. ChemPhysChem 16(5):960–969
Li L, Zhang M, Ronga M, Ruan W (2014) Studies on the transformation process of PVDF from α to β phase by stretching. RSC Adv 4:3938–3943
Zhang HP, Zhang P, Li ZH, Sun M, Wu YP (2007) A novel sandwiched membrane as polymer electrolyte for lithium ion battery. Electrochem Commun 9:1700–1703
Shen YJ, Reddy MJ, Chu PP (2004) Porous PVDF with LiClO4 Complex as ’solid’ and ’wet’ polymer electrolyte. Solid State Ionics 175:747–750
Angell CA, Liu C, Sanchez E (1993) Rubbery solid electrolytes with dominant cationic transport and high ambient conductivity. Nature 362:137–139
Ataollahi N, Ahmad A, Hamzah H, Rahman MYA, Mohamed NS (2013) Ionic conduction of blend poly (vinylidene fluoride-hexafluoro propylene) and poly (methyl methacrylate)-grafted natural rubber based solid polymer electrolyte. Int J Electrochem Sci 8:7875–7884
Ataollahi N, Ahmad A, Hamzah H, Rahman MYA, Mohamed NS (2012) Preparation and characterization of PVDF-HFP/MG49 based polymer blend electrolyte. Int J Electrochem Sci 7:6693–6703
Kumutha K, Alias Y, Said R (2005) FTIR and thermal studies of modified natural rubber based polymer electrolytes. Ionics 11:472–476
Idris R, Glasse MD, Latham RJ, Linford RG, Schlindwein WS (2001) Polymer electrolytes based on modified natural rubber for use in rechargeable lithium batteries. J Power Sources 94:206–211
Gray FM (1991) Solid Polymer Electrolyte–fundamentals and technological applications. Wiley-VCH, United Kingdom
Yu B, Zhou F, Wang C, Liu W (2007) A novel gel polymer electrolyte based on poly ionic liquid 1-ethyl 3-(2- methyacryloyloxy ethyl imidazolium iodide). Eur Polym J 43:2699–2707
Zhang C, Gamble S, Ainsworth D, Slawin AMZ, Andreev YG, Bruce PG (2009) Alkali metal crystalline polymer electrolytes. Nat Mater 8(7):580–584
Rajendran S, Sivakumar M, Subadevi R (2004) Investigations on the effect of various plasticizers in PVA-PMMA solid polymer electrolytes. Mater Lett 58:641–649
Ramesh S, Liew CW, Ramesh K (2011) Evaluation and investigation on the effect of ionic liquid onto PMMA-PVC gel polymer blend electrolytes. J Non-Cryst Solids 357:2132–2138
Saif AA, Jamal ZAZ, Sauli Z, Poopalan P (2011) Frequency dependent electrical properties of ferroelectric. Mater Sci 17(2):186–190
Ahmad Z, Isa MIN (2012) Ionics conduction via correlated barrier hoping mechanism in CMC–SA solid biopolymer electrolytes. Int J Latest Res Sci Technol 1(2):70–75
Ramesh S, Chai MF (2007) Conductivity, dielectric behavior and FTIR studies of high molecular weight poly (vinylchloride)–lithium triflate polymer electrolytes. Mater Sci Eng B 139:240–245
Ramesh S, Ong PL (2010) Effect of ethylene carbonate on the ionic conduction in poly (vinylidenefluoride-hexafluropropylene) based solid polymer electrolytes. Polym Chem 1:702–707
Ibrahim S, Mohd Yasin SM, Nee NM, Ahmad R, Johan MR (2012) Conductivity and dielectric behaviour of PEO-based solid nanocomposite polymer electrolytes. Solid State Commun 152(5):426–434
Pandey K, Singh M, Asthana N, Dwivedi MM, Agrawal SL (2011) Development of magnisio ferrite doped polymer electrolyte system for battery application. Int J Mater Sci 1:9–17
Pradhan DK, Choudhary RNP, Samantaray BK (2008) Studies of dielectric relaxation and AC conductivity behaviour of plasticized polymer nanocomposite electrolytes. Int J Electrochem Sci 3:597–608
Tripath SK, Gupta A, Kumari M (2012) Studies on electrical conductivity and dielectric behaviour of PVDF–HFP–PMMA–NaI polymer blend electrolyte. Bull Mater Sci 35(6):969–975
Su’ait MS, Ahmad A, Hamzah H, Rahman MYA (2011) Effect of lithium salt concentrations on blended 49% poly (methyl methacrylate) grafted natural rubber and poly (methyl methacrylate) based solid polymer electrolyte. Electrochim Acta 57:123–131
Eswaraiah V, Sankaranarayanan V, Ramaprabhu S (2001) Inorganic nanotubes reinforced polyvinylidene fluoride composites as low-cost electromagnetic interference shielding materials. Nanoscale Res Lett 6(1):137–147
Rajendran S, Sivakumar M, Subadevi R, Nirmala M (2004) Characterization of PVA–PVDF based solid polymer blend electrolytes. Phys B Condens Matter 348:73–78
Rajendran S, Mahendran O, Mahalingam T (2002) Thermal and ionic conductivity studies of plasticized pmma/pvdf blend polymer electrolytes. Eur Polym J 38(1):49–55
Zygadło-Monikowska E, Florjańczyk Z, Rogalska-Jońska E, Werbanowska A, Tomaszewska A, Langwald N, Greenbaum SG (2007) Lithium ion transport of solid electrolytes based on PEO/CF3SO3Li and aluminium carboxylate. J Power Sources 173:734–742
Rajendran S, Kannan R, Mahendran O (2001) An electrochemical investigation on PMMA-PVDF blend based polymer electrolytes. Mater Lett 49:172–179
Nallasamy P, Mohan S (2005) Vibrational Spectroscopic characterization of form II poly (vinylidene fluoride). Indian J Pure Appl Phys 43:821–827
Miao R, Liu B, Zhu Z, Liu Y, Li J, Wang X, Li Q (2008) PVDF-HFP-based porous polymer electrolyte membranes for lithium-ion batteries. J Power Sources 184(2):420–426
Acknowledgments
The authors are grateful to acknowledge the School of Chemical Sciences and Food Technology, Faculty of Science and Technology, National University of Malaysia, for allowing this research to be carried out. This work was funded by UKM under grant no. UKM-DLP-2012-021. We also would like to thank MOSTI for providing financial support under research grant of NND/NM (2)/TD11-046.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
TianKhoon, L., Ataollahi, N., Hassan, N.H. et al. Studies of porous solid polymeric electrolytes based on poly (vinylidene fluoride) and poly (methyl methacrylate) grafted natural rubber for applications in electrochemical devices. J Solid State Electrochem 20, 203–213 (2016). https://doi.org/10.1007/s10008-015-3017-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-015-3017-2