
Abstract
Enzyme-free amperometric ultrasensitive determination of hydrogen peroxide (H2O2) was investigated using a Prussian blue (PB) film-modified gold nanoparticles (AuNPs) graphite–wax composite electrode. A stable PB film was obtained on graphite surface through 2-aminoethanethiol (AET)-capped AuNPs by a simple approach. Field emission scanning electron microscope studies results in formation of PB nanoparticle in the size range of 60–80 nm. Surface modification of PB film on AET–AuNPs–GW composite electrode was confirmed by Fourier transform infrared attenuated total reflection (FTIR-ATR) spectroscopy studies. Highly sensitive determination of H2O2 at a peak potential of −0.10 V (vs. SCE) in 0.1 M KCl PBS, pH = 7.0) at a scan rate of 20 mVs−1 with a sensitivity of 23.58 μA/mM was observed with the modified electrode using cyclic voltammetry. The synergetic effect of PB film with AuNPs has resulted in a linear range of 0.05 to 7,800 μM with a detection limit of 0.015 μM for H2O2 detection with the present electrode. Chronoamperometric studies recorded for the successive additions of H2O2 with the modified electrode showed an excellent linearity (R 2 = 0.9932) in the range of 4.8 × 10−8 to 7.4 × 10−8 M with a limit of detection of 1.4 × 10−8 M. Selective determination of H2O2 in presence of various interferents was successfully demonstrated. Human urine samples and stain remover solutions were also investigated for H2O2 content.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrogen peroxide (H2O2) has gained much significance because it is the main byproduct of countless enzymatic reactions including glucose oxidase, urease, cholesterol oxidase, alcohol oxidase, sarcosine oxidase, galactose oxidase, and l-amino-acid oxidases. H2O2 is necessary for the metabolism of proteins, carbohydrates, fats, vitamins, and minerals. Besides its essential role for the production of estrogens, progesterone, and thyroxin in the body, H2O2 also helps to regulate blood sugar and cellular energy production. Oxidative damages in the body are caused by cellular H2O2 imbalance as it plays an important role in cell signaling and communication [1, 2]. H2O2 is also widely used in many fields, including pollution control, textile and paper bleaching, sterilization, and so on. Various methods have been developed for the determination of H2O2, such as spectrophotometry [3], fluorescence [4], chemiluminescence [5], and high-performance liquid chromatography [6]. Among electrochemical methods, amperometric biosensors based on electrodes modified with cyctochrome c [7, 8], horseradish peroxidase [9, 10], redox dyes [11, 12], hemoglobin [13, 14], and bienzymatic sensor [15] have been extensively studied. Even in the recent review [16], the authors have elaborated the various electrochemical sensing methods for determination of H2O2 using metal hexacyanoferrates (MHCFs), heme proteins with third generation biosensors, CNTs and graphene, metal and metal oxides, etc. However, there are several shortcomings in using enzymes such as high cost, low stability, and easy denaturation. Thus, it becomes necessary to fabricate enzyme-free electrochemical sensors for the detection of H2O2.
Self-assembled monolayers (SAMs) using bifunctional organo-thiols have offered a broad range of possibilities that can covalently anchor nanomaterials with one end and redox mediator on the other end [17–19] which finds wide applications in electroanalysis. SAMs of thiols with nanomaterials provides a simple, rapid, and convenient system to modulate in particular the electronic properties of metals, metal oxides, and semiconductors [20, 21]. Currently, there is a growing interest in use of metal nanoparticles to successfully modify electrode surfaces for both chemical sensor and biosensor applications. Gold nanoparticles (AuNPs) have been used in electrocatalysis for numerous biosensor applications because of its high stability, excellent electronic property, and complete recovery during biochemical applications [22]. High cost, denaturation, and non-reliability in performance of successive analysis using enzymes have led to development of numerous enzyme-free biosensors for determination of H2O2 using AuNPs. Few among them are hemoglobin (Hb)/gold colloid (nano-Au)/l-cysteine(l-cys)/nano-Au/nanoparticles Pt (nano-Pt)–chitosan (CHIT) composite film-modified platinum disk electrode [23], poly (diallyldimethylammonium chloride) colloidal carbon sphere silica with gold nanoparticles (AuNPs–C@SiO2)–Hb composite electrode [24], poly-brilliant cresyl blue (Poly(BCB))/gold nanoparticles (Au–NPs)-modified electrode [25], Nafion/myoglobin/colloidal gold nanoparticles/GCE [26], Hb/nano-Au/thionine/2,6-pyridinedicarboxylic acid (PDC) /GCE [19], gold nanoparticles (GNPs) on ordered mesoporous carbon (OMC) electrode [27], Fe(III)/MPBA/3D-Au [28], Nanoporous gold as non-enzymatic sensor [29], and Azure-A/gold NCs-modified electrode [30]. These reports on AuNPs-modified enzyme-free biosensor have enhanced the sensitivity and detection limits for the determination of H2O2 in comparison with the bare electrode.
PB analogues are a class of mixed valence hexacyanoferrate compounds which are known to exhibit interesting electrochemical, photochemical, biochemical, and magnetic properties [31–34]. PB analogues are being denoted as “artificial peroxidases” which can exhibit not only rapid catalytic activity toward reduction of hydrogen peroxide but also lowers the overpotential [35]. Hence, PB analogues have been widely used as electron transfer mediators in the oxidase-based amperometric biosensors [36] that can eliminate the interference from the coexisting substances such as ascorbic acid (AA), paracetomol, and uric acid (UA). MHCF-modified electrodes for the determination of H2O2 reported so far are titanium dioxide electrode modified with potassium hexacyanoferrate (III) [37], vanadium hexacyanoferrate [38], PB on a carbon ionic liquid electrode [39], chromium (III) hexacyanoferrate (II)-modified glassy carbon electrode [40], CoHCF and CuHCF film-modified electrode [41], various MHCF polymethylmetacrylate-modified graphite electrode [42], and ruthenium oxide hexacyanoferrate electrode [43].
In the present work, we have developed a novel Prussian blue film modification on 2-aminoethanethiol (AET)-capped AuNPs on graphite–wax (GW) electrode (herein after called as PB film-modified electrode) for the enzyme-free determination of H2O2. PB film was characterized using field emission scanning electron microscopy (FESEM) and Fourier transform infrared attenuated total reflection (FTIR-ATR) spectroscopy. Electrochemical behavior of PB film-modified electrode was characterized by cyclic voltammetry (CV), differential pulse voltammetry (DPV), and chronoamperometric studies. In addition, the utility of the proposed biosensor was also investigated for the detection of H2O2 from human urine samples and laundry stain remover solutions.
Experimental section
Chemicals
Hydrogen peroxide (H2O2) was obtained from Merck, India. Graphite powder (≈1–2 μm) was purchased from Aldrich Chemicals, Germany. Chloroauric acid (HAuCl4), AET, trisodium citrate, and all other reagents employed were of analytical grade and used as received. H2O2 (0.1 M) was prepared using potassium dihydrogen phosphate buffer solution (PBS) (0.1 M, pH = 7.0). AET solution (20 mM) was prepared in dry ethanol. Potassium nitrate (0.1 M) and potassium ferrocyanide (0.02 M) solutions were prepared in double distilled (DD) water.
Apparatus
Cyclic voltammetry experiments were carried out with an electrochemical workstation CHI 660B (CH Instruments, USA) and the output was recorded using desktop computer. A cell of volume of 75 ml with standard calomel as reference electrode, PB film-modified electrode as working electrode and platinum electrode as the auxiliary electrode was used throughout the electrochemical measurements. UV–vis characterization for the synthesized AuNPs was carried out by a fiber optic spectrometer (Ocean optics, Inc. Florida, USA) with deuterium–tungsten source. FESEM image of the PB film-modified electrode were obtained using FESEM (Hitachi SU6600) at an accelerating voltage of 15 kV. FTIR-ATR spectroscopy studies of the PB film modification was carried out using Bruker tensor 27 model. The experiments were carried out after purging the electrolyte solutions with pure nitrogen to ensure the solution is free from atmospheric oxygen.
Preparation of PB film-modified electrode
The PB film-modified electrode was constructed using our previous procedure [44] with a slight modification: trisodium citrate-stabilized AuNP was prepared using sodium borohydride reduction method [45]. Spectroscopic graphite powder of (2 g) was stirred with an optimized volume of 60 mL of AuNPs colloidal solution (12 μM) for period of 2 h. After centrifugation, the AuNPs–graphite composite powder was dried at room temperature. The AuNPs-adsorbed graphite powder was further mixed thoroughly and stirred with an optimized volume of 60 mL of 20 mM ethanolic solution of AET for 2 h, centrifuged, and dried at room temperature. The resultant AET–AuNPs–graphite composite was mixed with paraffin wax thoroughly in the ratio of 4:1 at warm condition. With the help of a 3-mm diameter glass tube, the AET–AuNPs–GW composite powder was packed tightly into the glass tube and the composite electrode was gently pushed out of the test tube immediately. After drying, one end of composite electrode surface was polished and then derivatized using potassium ferrocyanide (0.02 M) present in potassium nitrate solution (0.1 M, pH = 6.0) by cycling the potential 30 times between −0.2 and 1.0 V at the sweep rate of 50 mV s−1. Finally after derivatization, as prepared PB film-modified electrode was washed thoroughly with double distilled (DD) water. In addition, surface modification of PB film without AuNPs was carried out with AET–graphite–wax composite to compare and investigate the synergic effect of PB film with the AuNPs over the electrochemical performance of PB film-modified electrode.
Results and discussion
Physical characterization studies
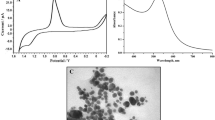
A preliminary investigation with UV–vis spectra and FESEM imaging (not shown) has confirmed the formation of AuNPs in the range of 15–30 nm as reported in our earlier article [46]. PB film modification on the AET–AuNPs–GW composite has resulted in PB particles formation in the range of 60–80 nm as shown in the FESEM image (Fig. 1A). Curves a and b of Fig. 1B corresponds to the FTIR-ATR images of bare AET–AuNPs–GW composite and PB film-modified electrode, respectively. A sharp peak around 2,100 cm−1 (curve b) is the characteristic stretching frequency of –C≡N ligand in [Fe(CN)6]4− anion present in the PB film which is in full agreement with the literature data [47].

A FESEM image of PB film; B FTIR-ATR spectra of (a) AET–AuNPs–GW electrode and (b) PB film-modified electrode
Electrochemical behavior of PB film-modified electrode
Participation of alkali cation of the background electrolyte during the redox reactions of high-spin and low-spin transitions of Fe2+/3+ of the PB film is determined by the size of cation which moves in and out of the lattice and will in turn decide the electrochemical behavior of the PB film-modified electrode. The effect of metal ions such as K+, Na+, NH4 +, and Li+ towards the electrochemical behavior of the PB film-modified electrode was investigated in the potential range of 1.0 to −0.2 V (negative scan). It was observed that the Na+, Li+, and NH4 + ions showed a very poor redox behavior for the PB film-modified electrode. Among all these ions, only K+ ion was found to exhibit an excellent redox behavior which gives rise to two pair of well-resolved redox peaks at formal potentials of 0.12 and 0.80 V in presence of 0.1 M KCl at the scan rate of 20 mVs−1 which corresponds to the high-spin (peaks I and I′) and low-spin (peaks II and II′) transitions of FeII/III redox reactions of PB film, respectively. Effect of anions had a very negligible effect on the redox process of the PB film as shown in Fig. 2A. With increasing scan rate, the cathodic and anodic peak currents of both the redox couples Fe II/III of the PB film increased linearly in the scan range of 10–150 mVs−1 in 0.1 M KCl as shown in the (Fig. 2B). The increase in peak currents of both the high and low-spin transitions of FeII/III with respect to square root of scan rates as shown in the inset of Fig. 2B suggests the electrochemical behavior of the PB film-modified electrode is purely governed by diffusion-controlled process.

A CVs of PB film-modified electrode in presence of 0.1 M of K2SO4, KCl, KNO3, LiCl, NaCl, and NH4Cl at the scan rate of 20 mV/s. B CVs of PB film-modified electrode at different scan rates, from inside to outer are 10–150 mV/s with increments of 10 mV/s in 0.1 M KCl. The insets a and b show the dependence of peak current I pa and I pc on square root of scan rate (ν)
Figure 3 shows the cyclic voltammograms of bare graphite wax electrode (curve a), PB film-modified graphite–wax electrode without AuNPs (curve b) and with AuNPs (curve c).
From the figure, it is observed that the synergic behavior of PB film with the AuNPs has facilitated the redox activity of PB film and enhanced the peak current by fourfold with excellent stability in comparison with the modified electrode without AuNPs. The PB film-modified electrode with AuNPs was used for further investigations.

CVs of (a) bare composite, PB film-modified electrode (b) without AuNPs, and (c) with AuNPs in 0.1 M KCl solution; scan rate: 20 mV/s
Electrocatalytic reduction of H2O2 at PB film-modified electrode
To investigate the electrocatalytic reduction of H2O2 the potential window of the PB film-modified electrode was fixed between 0.6 and −0.3 V (negative scan). The high-spin transition of FeII/III system of the PB film facilitates the electrocatalytic reduction of H2O2. Figure 4 shows the CVs response of the PB film-modified electrode for the successive additions of H2O2 in 0.1 M KCl (PBS, pH = 7). The reduction current of H2O2 starts at a potential 0.12 V and reaches a maximum current at −0.10 V with increase in background current at the tail. The range of detection for H2O2 at the PB film-modified electrode was observed between 0.05 and 7,800 μM and the limit of detection was found to be 0.015 μM with a very high sensitivity of 23.58 μA/mM of H2O2. The effect of pH on the PB film-modified electrode towards the catalytic reduction of H2O2 was studied and it was found that the electrode is active in the pH range of 2–9 (figure not included) and a maximum current was observed at the neutral pH (pH = 7). In comparison with earlier report on reduction of H2O2 [40, 48], the present method exhibits profound increase in the peak current with a very high sensitivity. From the calibration plot shown in the inset of Fig. 4, it is observed that the catalytic current increases linearly with increase in concentration of H2O2 (R 2 = 0.9941). The mechanism of electrocatalytic reduction of H2O2 at the PB film-modified electrode is given as follows [39]:

CV response of PB film-modified electrode (a) in absence and (b–f) in presence of successive additions of H2O2 in 0.1 M KCl (PBS, pH = 7.0) at the scan rate of 20 mVs−1. The inset shows the calibration plot of current response versus concentration of H2O2
Highly sensitive technique such as DPVs was also employed to study the PB film-modified electrode performance towards successive additions of H2O2 and the result is shown in Fig. 5. It is observed from the DPV curves, similar to CV curves, the reduction current starts well before 0.12 V (vs. SCE) and increases further to attain a maximum current at −0.10 V. The overlapping peaks are observed at +0.10 and −0.10 V in higher concentrations of H2O2 still the initial peak around +0.10 V corresponds to redox wave of PB film which is significant. The direct reduction of H2O2 at the bare electrode cannot be completely excluded and this observation on DPVs is quiet peculiar.

DPV curves of PB film-modified electrode for successive additions of H2O2 in 0.1 M KCl (PBS, pH = 7.0) at the scan rate of 20 mVs−1
The AuNPs microenvironment in the graphite–wax composite has offered a stable PB film modification with an enhanced sensitivity towards the determination of H2O2. From Table 1, it is understood that the results of present method based on AuNPs and PB film-modified enzyme-free electrodes is comparable with reported methods. The entire PB film modification and its electrocatalytic reduction towards H2O2 are illustrated in the Scheme 1. The reproducibility of the PB film-modified electrode was investigated by determining the 0.5 mM of H2O2 in PBS (pH 7.0) for ten successive assays which has shown a relative standard deviation (R.S.D = 1.3 %). The fabrication reproducibility for three similar PB film-modified electrodes gave an R.S.D. of 1.5 % for the determination of 0.5 mM H2O2. These results suggest that the proposed electrode exhibits a good stability and reproducibility, which can be satisfactorily applied for the determination of H2O2 in commercial samples.

Surface modification of PB film-modified electrode and its reduction mechanism towards H2O2
Chronoamperometry determination of H2O2
The analytical applicability of the PB film-modified electrode towards the determination of H2O2 in flow systems was also investigated. From the hydrodynamic plot of potential versus catalytic current (not included), it was observed that the reduction current starts well before 0.12 V and increases slowly and attains a maximum at −0.10 V. Hence, an applied potential of −0.10 V was fixed as the working potential for the amperometric determination of H2O2. Figure 6 shows the chronoamperometric response of PB film-modified electrode for the successive additions of 0.1 ml of 0.1 M of H2O2 in the time interval of 30 s at a fixed potential of −0.10 V in 75 ml of 0.1 M KCl (PBS, pH = 7.0) at the scan rate of 20 mVs−1. From the inset of Fig. 6, it is evident that PB film-modified electrode exhibits an excellent performance even at flow system with a good linearity (R 2 = 0.9932) in the range of 4.8 × 10−8 to 7.4 × 10−8 M with a detection limit of 1.4 × 10−8 M of H2O2.

Chronoamperometric responses of the PB film-modified electrode for the successive additions of 0.1 ml of 0.1 M stock solution of H2O2 in 75 ml of 0.1 M KCl (PBS, pH = 7.0) at the applied potential of −0.10 V with a stirring rate of 300 rpm. Inset shows the calibration plot for H2O2
Selectivity of the PB film-modified electrode
Selective determination of H2O2 is an important factor for the performance of the proposed sensor. High concentrations (1 mM) of the main interfering substances such as AA, dopamine (DA), UA, l-cysteine (l-Cys), sodium sulfite (SO3 2−), and sodium nitrite (NO2 −) were investigated using chronoamperometric technique to evaluate the selectivity of the sensor towards detection of H2O2. Amperometric response for the detection of H2O2 at PB film-modified electrode in presence of above-mentioned interferents in 0.1 M KCl (PBS, pH = 7.0) is shown in the (Fig. 7). It is observed from the figure that there is negligible effect by all these interferents against the detection of H2O2.

Amperometric response of the PB film-modified electrode towards interference effect for the determination of H2O2 in presence of various interferents in 0.1 M KCl (PBS, pH = 7.0) at a peak potential of −0.10 V with a stirring rate of 300 rpm
H2O2 determination from urine samples and stain remover solutions
The proposed electrode was also investigated for the detection of H2O2 in human urine samples and commercially available peroxide stain remover solutions. Three different commercial stain remover solutions were purchased from a local supermarket. From the above samples, 10 μl was injected into the 0.1 M PBS solution followed by addition of standard H2O2 solutions. The cyclic voltammetry responses (not shown) were recorded using the PB film-modified electrode. The above experiment was repeated for seven times and the analytical data obtained are presented in the Table 2. Commercially available peroxide solutions do contain a small amount of organic stabilizer to minimize the decomposition of H2O2. Amino trimethylene phosphonic acid or hydroxyl ethylidene diphosphonic acid is the typical organic stabilizer used to prevent the decomposition of H2O2 [49]. But practically, these organic stabilizers do not interfere the peroxide determination by electrochemical method. In addition, human urine sample was diluted in the ratio of 1:100 times with DD water and used as such for analysis. Real sample analysis was performed with the above human urine sample by standard addition method and the recovery data are also tabulated as shown in Table 2.
Conclusion
In summary, a novel PB film modification was carried out successfully on AET–AuNPs–GW composite for the enzyme-free sensing of H2O2. The intrinsic structure stability of PB film and the large surface area of AuNPs have undergone a synergistic process in order to improve the analytical performance and reliability of PB film-modified electrode for the determination of H2O2. The proposed PB film-modified electrode has shown a low detection limit of 15 nM with a very good sensitivity of 23.58 μA/(H2O2) mM in 0.1 M KCl. The interference effect towards the electrocatalytic response of the PB film-modified electrode for the reduction of H2O2 in presence of AA, DA, UA, l-Cys, SO3 2−, and NO2 − ions was found to be insignificant. Moreover, the present method has exhibited an excellent recovery for the real sample analysis of H2O2 in both human urine samples and commercial stain remover solutions.
References
Dröge W (2007) Free radicals in the physciological control of cell function. Physiol Rev 81:47–95
Xu J, Shang F, Luong JHT, Razeeb KM, Glennon JD (2010) Direct electrochemistry of horseradish peroxidase immobilized on a monolayer modified nanowire array electrode. Biosens Bioelectron 25:1313–1318
Guo ZX, Shen HX, Li L (1999) Spectrophotometric determination of hydrogen peroxide and glucose based on hemin peroxidase-like catalyzed oxidation of bromopyrogallol red. Microchim Acta 131:171–176
Albers AE, Okreglak VS, Chang CJ (2006) A FRET-based approach to ratiometric fluorescence detection of hydrogen peroxide. J Am Chem Soc 128:9640–9641
Lei W, Duerkop A, Lin Z, Wu M, Wolfbeis OS (2003) Detection of hydrogen peroxide in river water via a microplate luminescence assay with time-resolved (“Gated”) detection. Microchim Acta 143:269–274
Pinkernell U, Effkemann S, Karst U (1997) Simultaneous HPLC determination of peroxyacetic acid and hydrogen peroxide. Anal Chem 69:3623–3627
Xiang C, Zou Y, Sun L, Xu F (2008) Direct electron transfer of cytochrome c and its biosensor based on gold nanoparticles/room temperature ionic liquid/carbon nanotubes composite film. Electrochem Commun 10:38–41
Jiang X, Zhang L, Dong S (2006) Assemble of poly(aniline-co-o-aminobenzenesulfonic acid) three dimensional tubal net-works onto ITO electrode and its application for the direct electrochemistry and electrocatalytic behavior of cytochrome c. Electrochem Commun 8:1137–1141
Mao L, Yamamoto K (2000) Glucose and choline on-line biosensors based on electropolymerized Meldola's blue. Talanta 51:187–195
Yang Y, Yang G, Huang Y, Bai H, Lu X (2009) A new hydrogen peroxide biosensor based on gold nanoelectrode ensembles/multiwalled carbon nanotubes/chitosan film-modified electrode. Colloids Surf A Physicochem Eng Aspects 340:50–55
Shobha Jeykumari DR, Narayanan SS (2007) Covalent modification of multiwalled carbon nanotubes with neutral red for the fabrication of an amperometric hydrogen peroxide sensor. Nanotechnology 18:125501
Siao HS, Chen SM, Lin KC (2011) Electrochemical study of PEDOT-PSS-MDB- modified electrode and its electrocatalytic sensing of hydrogen peroxide. J Solid State Electrochem 15:1121–1128
Shi G, Sun Z, Liu M, Zhang L, Liu L, Qu Y, Jin L (2007) Electrochemistry and electrocatalytic properties of hemoglobin in layer-in-layer films of SiO2 with vapour–surface sol–gel deposition. Anal Chem 79:3581–3588
Liu H, Rusling JF, Hu N (2004) Electroactive core-shell nanocluster films of heme proteins, polyelectrolytes, and silica nanoparticles. Langmuir 20:10700–10705
Shobha Jeykumari DR, Narayanan SS (2008) Fabrication of bienzymatic nanobiocomposite electrode using functionalized carbon nanotubes for biosensing applications. Biosens Bioelectron 23:1686–1693
Chen W, Cai S, Ren QQ, Wen W, Zhao YD (2012) Recent advances in electrochemical sensing of hydrogen peroxide. Analyst 137:49–58
Ma L, Yuan R, Chai Y, Chen S, Ling S (2009) Amperometric hydrogen peroxide biosensor based on covalently immobilizing thionine as a mediator. Bioprocess Biosyst Eng 32:537–544
Lopez JA, Manrioquez J, Mendoza S, Godionez LA (2007) Design and construction of nickel hexacyanoferrate—starburst PAMAM dendrimer modified gold electrodes for the potentiometric detection of potassium in aqueous media. Electrochem Commun 9:2133–2139
Tao W, Liu Y, Pan D, Nie L, Yao S (2004) Investigation of the effects of Au-colloid modification on cobalt hexacyanoferrate film growth and mass transport by electrochemical quartz crystal microbalance. J Colloids Interface Sci 275:257–263
Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM (2005) Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem Rev 105:1103–1169
Vericat C, Vela ME, Benitez GA, Martin Gago JA, Torrelles X, Salvarezza RC (2006) Surface characterization of sulfur and alkanethiol self-assembled monolayers of Au(111). J Phys Condens Matter 18:867–900
Pavlov V, Xiao Y, Willner I (2005) Inhibition of the acetycholine esterase-stimulated growth of Au nanoparticles: nanotechnology-based sensing of nerve gases. Nano Lett 5:649–653
Yang G, Yuan R, Chai YQ (2008) A high-sensitive amperometric hydrogen peroxide biosensor based on the immobilization of hemoglobin on gold colloid/l-cysteine/gold colloid/nanoparticles Pt–chitosan composite film-modified platinum disk electrode. Colloids Surf B Biointerfaces 61:93–100
Wang Y, Chen X, Zhu JJ (2009) Fabrication of a novel hydrogen peroxide biosensor based on the AuNPs–C@SiO2 composite. Electrochem Commun 11:323–326
Ashok Kumar S, Wang SF, Chang YT (2010) Poly (BCB)/Au-nanoparticles hybrid film modified electrode: preparation, characterization and its application as a non-enzymatic sensor. Thin Solid Films 518:5832–5838
Yang W, Li Y, Bai Y, Sun C (2006) Hydrogen peroxide biosensor based on myoglobin/colloidal gold nanoparticles immobilized on glassy carbon electrode by a Nafion film. Sens Actuators B 115:42–48
Wang L, Bo X, Bai J, Zhu L, Guo L (2010) Gold nanoparticles electrodeposited on ordered mesoporous carbon as an enhanced material for nonenzymatic hydrogen peroxide sensor. Electroanalysis 22:2536–2542
Zheng M, Li P, Yang C, Zhu H, Chen Y, Tang Y, Zhou Y, Lu T (2012) Ferric ion immobilized on three-dimensional nanoporous gold films modified with self-assembled monolayers for electrochemical detection of hydrogen peroxide. Analyst 137:1182–1189
Meng H, Yan X, Liu J, Gu J, Zhou Z (2011) Nanoporous gold as non-enzymatic sensor for hydrogen peroxide. Electrochim Acta 56:4657–4662
Priya C, Sivasankari G, Narayanan SS (2012) Electrochemical behaviour of Azure A/gold nanoclusters modified electrode and its application as non-enzymatic hydrogen peroxide sensor. Colloids Surf B Biointerfaces 97:90–96
Shen X, Wu S, Liu Y, Wang K, Xu Z, Liu W (2009) Morphology syntheses and properties of well- defined Prussian blue nanocrystals by a facile solution approach. J Colloids Interface Sci 329(188):195
Hornok V, Dekany I (2007) Synthesis and stabilization of Prussian blue nanoparticles and application for sensors. J Colloids Interface Sci 309:176–182
Narayanan SS, Scholz F (1999) A comparative study of electrocatalytic activities of some metal hexacyanoferrates for the oxidation of hydrazine. Electroanalysis 11:465–469
Araminaite R, Garjonyte R, Malinauskas A (2010) Electrocatalyic reduction of hydrogen perioxide at Prussian blue modified electrodes: a RDE study. J Solid State Electrochem 14:149–155
Dostal A, Meyer B, Scholz F, Schroder U (1995) Electrochemical study of microcrystalline solid Prussian blue particles mechanically attached to graphite and gold electrodes: electrochemically induced lattice reconstruction. J Phys Chem 95:2096–2103
Karyakin AA, Karyakina E, Gorton L (1998) Amperometric biosensor for glutamate using Prussian blue-based artificial perioxdase as a transducer for hydrogen peroxide. J Electroanal Chem 456:97–104
Mishima Y, Motonaka J, Maruyama K, Ikeda S (1998) Determination of hydrogen peroxide using a potassium hexacyanoferrate(III) modified titanium dioxide electrode. Analytica Chim Acta 358:291–296
Tsiafoulis CG, Trikalitis PN, Prodromidis MI (2005) Synthesis, characterization and performance of vanadium hexacyanoferrate as electrocatalyst of H2O2. Electrochem Commun 7:1398–1404
Li Y, Liu X, Zeng X, Liu Y, Liu X, Wei W, Luo S (2009) Nonenzymatic hydrogen peroxide sensor based on a Prussian blue-modified carbon ionic liquid electrode. Microchim Acta 165:393–398
Lin MS, Tseng TF, Shih WC (1998) Chromium (III) hexacyanoferrate (II)-based chemical sensor for the cathodic determination of hydrogen peroxide. Analyst 123:159–163
Pauliukaite R, Florescu M, Christopher Brett MA (2005) Characterization of cobalt and copper hexacyanoferrate-modified carbon film electrodes for redox-mediated biosensors. J Solid State Electrochem 9:354–362
Gonzalez GLL, Kahlert H, Scholz F (2007) Catalytic reduction of hydrogen peroxide at metal hexacyanoferrate composite electrodes and applications in enzymatic analysis. Electrochim Acta 52:1968–1974
Thiago RL, Paixao C, Bertotti M (2008) Ruthenium oxide hexacyanoferrate modified electrode for hydrogen peroxide detection. Electroanalysis 20:1671–1677
Prabhu P, Suresh Babu R, Narayanan SS (2011) Amperometric determination of l-dopa by nickel hexacyanoferrate film modified gold nanoparticle graphite composite electrode. Sens Actuators B 156:606–614
Brown KR, Walter DG, Natan MJ (2000) Seeding of colloidal Au nanoparticle solutions improved control of particle size and shape. Chem Mater 12:306–313
Prabhu P, Suresh Babu R, Narayanan SS (2011) Electrocatalytic oxidation of l-tryptophan using copper hexacyanoferrate film modified gold nanoparticle graphite-wax electrode. Colloids Surf B Biointerfaces 87:103–108
Sheng QL, Yu H, Zheng JB (2007) Sol–gel derived carbon ceramic electrode for the investigation of electrochemical behavior and electrocatalytic activity of neodymium hexacyanoferrate. Electrochim Acta 52:4506–4512
Lin MS, Shih WC (1999) Chromium hexacyanoferrate based glucose biosensor. Analytica Chim Acta 381:183–189
Kumar SA, Chen SM (2007) Direct electrochemistry and electrocatalysis of myoglobin on redox-active self-assembling monolayers derived from nitroaniline modified electrode. Biosens Bioelectron 22:3042–3050
Acknowledgements
The authors acknowledge the financial assistance from University Grants Commission (UGC), New Delhi, India, and Department of Science and Technology for PURSE program in support of this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Prabhu, P., Babu, R.S. & Narayanan, S.S. Synergetic effect of Prussian blue film with gold nanoparticle graphite–wax composite electrode for the enzyme-free ultrasensitive hydrogen peroxide sensor. J Solid State Electrochem 18, 883–891 (2014). https://doi.org/10.1007/s10008-013-2288-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-013-2288-8