
Abstract
A convenient method for the production of graphene is developed using the electrochemical reduction of graphite oxide (GO) in solution without assembling it onto the electrode. The samples were examined by X-ray diffraction, X-ray photoelectron spectroscopy, Fourier transform infrared spectroscopy, and Raman spectroscopy. The results show that the number of oxygen functional groups can be significantly decreased. The electrochemical capacitance of the prepared graphene after 8 h of reduction is 158.5 F g−1 at 0.5 A g−1, much higher than that of GO and carbon nanotubes. The mechanism for this reaction is also proposed in this paper.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since reported in 2004 [1], graphene has attracted great attention due to its unique electronic, thermal, and mechanical properties [2–4]. Many routes have been developed to synthesize the two-dimensional graphene sheets such as chemical vapor deposition of hydrocarbons on metal substrate [5] and thermal decomposition [6]. However, it is still unfeasible to fabricate graphene on a mass without using high temperature, inert atmospheres, and complex instruments [7]. Although chemical method is an effective approach to change graphite oxide (GO) into graphene on a large scale, the reducing agents such as hydrazine and dimethylhydrazine are very hazardous and toxic and can break graphene into pieces. Therefore, developing a safe and environmentally friendly way of producing graphene is becoming more and more necessary. Dreyer et al. [8] have reported the reduction of GO by alcohols at 100 °C. Nethravathi and Rajamathi [7] have reduced graphene by water, ethanol, 1-butanol, and ethylene glycol at about 200 °C. Fan et al. [9] have found that graphene can be prepared by heating an exfoliated GO suspension under strong alkaline conditions at 50–90 °C. These methods still require high temperature and a long time of preparation. Recently, electrochemical reduction of GO by using cyclic voltammetry has been introduced [10–12]. Electrochemistry is a green method for preparing graphene without using a reducing agent and high temperature. The ionic-liquid-functionalized graphene sheets are synthesized from graphite through a mild electrochemical approach [13]. By assembling GO onto a gold electrode, the graphene films can be obtained using electrochemical reduction at room temperature [14]. An et al. [15] have reduced the GO film on a silicon electrode by using an electrophoretic deposition process. To date, the conventional electrochemical synthesis of graphene is carried out after the GO is assembled on the electrode. However, the quantity of graphene obtained through conventional electrochemical reduction using an electrode with a limited area to perform the reaction by one experiment was confined to a few tens of micrograms, leading to low-yield production. Moreover, the produced graphene may be limited to the surface of electrode because the underplayed parts not in touch with the solution could not easily be reduced. To date, no research regarding the electrochemical reduction of GO without assembling the GO powder or film onto the electrode has been reported. In this paper, we present a new electrochemical method to prepare graphene directly in solution with a self-prepared electrochemical cell containing a large effective reaction surface area. This method performs a green, high-yield, low-cost, fast graphene production, which may have potential application in commercialization.
Experimental
Synthesis of GO
GO was prepared and purified according to the Hummers method [16].
Pretreatment of multi-walled carbon nanotubes
Carbon nanotubes (CNTs) used were produced using the chemical vapor deposition method. The CNTs were refluxed in 4.0 M HNO3 at 120 °C for 8 h and then extensively washed with deionized water.
Electrochemical reduction of GO
The new electrochemical cell for the scale-up synthesis as shown in Fig. 1 consists of a graphite cylinder (10 cm in diameter) with a graphite cover which is embedded with 10–14 graphite electrodes (each is about 0.3 cm in diameter) that act as cathodic working electrodes. The platinum plate serves as the anodic electrode. After 1 g of as-synthesized GO powder was scattered in the cell, the electrochemical reduction was carried out with the solution being stirred at the same time. The electrolyte solution is 0.1 M KNO3. The electrochemical reduction was carried out at an applied direct current voltage of 20 V with the use of a WYK-1502 power source. The reducing time was in the range of 10 min to 24 h.

Diagram of the electrochemical graphite cell serving as the cathode
Characterization
X-ray diffraction (XRD) data of the samples were collected by a Bruker D8 ADVANCE diffractometer using CuKα radiation (λ = 0.15406 nm). The 2θ angular regions between 5° and 70° were explored at a scan rate of 5°/min. The Fourier transform infrared spectroscopy (FT-IR) spectra of GO and electrochemically reduced graphite oxide (ER-GO) were characterized by a Thermo Nicolet NEXUS spectrometer. The samples (0.5 wt%) were mixed and grounded uniformly with monomorph KBr powder (99.5 wt%) in an agate mortar, and the powder was then pressed into disc specimens with an oil hydraulic press. The FT-IR spectrometer was operated under a transmittance mode. The samples were also analyzed by Raman spectroscopy (HORIBA Jobin Yvon, Ar+ laser emitter with a wavelength of 514.5 nm). X-ray photoelectron spectroscopy (XPS) measurement was performed by an ESCALAB MK II apparatus using non-monochromated MgKα radiation at 300 W. The instrument resolution was 0.75 eV. Thermogravimetric analysis (TGA) of GO and ER-GO was done in a STA 409 PC simultaneous thermal analyzer. The weight loss of the samples was monitored from room temperature to 700 °C at a heating rate of 10 °C min−1 in nitrogen atmosphere. The features of the sample were observed with high resolution transmission electron microscopy (HR-TEM; JEM-2100). Electrochemical experiments were performed by a CHI 750 electrochemical workstation (Chenhua, Shanghai). The electrochemical measurements were made with a three-electrode cell at 25 °C. Pt foil and saturated calomel electrode were used as the counter and reference electrodes, respectively.
Results and discussion
As shown in Fig. 2, the yellow-brown exfoliated GO suspension quickly turned into brown black after 10 min of electrochemical reduction. The suspension turned homogeneous black after 1 h. After it was left still for one night, the black sediment appeared at the bottom of beaker. These results revealed that GO had undergone fast deoxygenation by electrochemical reduction.

Images of the exfoliated GO suspension before and after electrochemical reduction with a reduction time of 20 min, 30 min, and 1 h in 0.1 M KNO3
In Fig. 3, the microstructure of GO and ER-GO nanosheets obtained by 8 h of reduction, which were analyzed by TEM observation, is shown. Figure 3a shows a TEM image of GO, clearly illustrating the flake-like shapes of GO. Selected area electron diffraction (SAED) pattern confirmed the disordered nature of the GO. Only diffraction rings were observed in the SAED pattern of GO, indicating that GO is amorphous. After exposure under the electron beam for a few minutes during the TEM observation, the GO was found unstable and tended to be broken. Like the crumpled silk veil waves, the observed large transparent graphene sheets, however, are very stable under the electron beam. The crystalline structure of the graphene obtained by electrochemical reduction was evident because of the defined diffraction spots. As shown in the figure insert of the SAED pattern, the graphitic laminar structure can be observed in the ordered region.

TEM images of GO (a) and ER-GO (b)
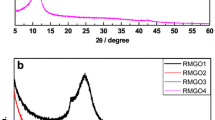
The XRD patterns of the as-prepared GO and ER-GO with different electrochemical reduction times are shown in Fig. 4. The GO powder shows a larger interlayer spacing than that of graphite with a major peak at 11.9° corresponding to the interlayer spacing of 7.43 Å due to the oxygen functional groups and water molecules in the interlayer of hydrophilic GO. Compared with the GO, the ER-GO samples show a very broad 002 reflection, while the peak of 001 at a 2θ value of about 11.9° decreases obviously as the reduction time increases. The major peak of ER-GO is observed at 2θ values of 24.9°, 25.6°, and 25.8° with a basal spacing of 3.57, 3.48, and 3.46 Å, respectively, which are higher than those of well-ordered graphite having a basal spacing of 3.35 Å. These results confirm that the oxygen and hydrogen causing the basal spacing of GO can be effectively removed with the increase of the reducing time. The broad 002 reflection of ER-GO indicates that the powder contains largely free graphene sheets in poor order along the stacking direction [7].

XRD patterns of GO and ER-GO with a reduction time of 10 min, 1 h, and 6 h
Figure 5 shows the IR spectra of GO and ER-GO. As for GO, a strong and broad absorption observed at about 3,400 cm−1 can be ascribed to the O–H stretching vibration. The peak at 1,726 cm−1 can be assigned as the stretching of C=O of COOH, and the peak at 1,622 cm−1 can be attributed to the O–H bending vibration, epoxide groups, and skeletal ring vibrations [7]. The peak of the tertiary C–OH groups is located at 1,393 cm−1, and the C–OH and C–O stretching peaks were at 1,220 and 1,050 cm−1, respectively [17]. After electrochemical reduction for 10 min, the ER-GO peak intensity of O–H stretching vibration at about 3,400 cm−1 presents an obvious decrease, while the peaks at 1,726 and 1,622 cm−1 cannot be observed. The skeletal vibration of the graphene sheets appears at 1,560 cm−1 [7]. The peaks at 1,220 and 1,050 cm−1 decrease obviously. These indicate that the hydroxyl and epoxide can be significantly reduced by this electrochemical method after 10 min.

IR spectra of GO and ER-GO with a reduction time of 10 min
Raman spectroscopy is commonly taken as an effective method to fabricate the reduction of GO. Figure 6 shows the Raman spectra of GO and ER-GO using an excitation wavelength of 514.5 nm. All measurements were carried out in powder form which was deposited on a silicon wafer in the absence of a solvent. GO displays two bands at ∼1,351 and ∼1,599 cm−1, corresponding to D and G modes, respectively. The D band is due to the defects or edges [18]. Higher disorder leads to a broader G band as well as D band with relatively higher intensity compared to that of G band [19]. The reduction of GO results in the crystalline order of sp2 carbon atoms with an increasing π-electron conjugation sp2 network [11]. Consequently, the G band associated with a graphite lattice will shift to a lower value. As can be seen from Fig. 6, the G band, lower than that of GO, shifts from 1,576 to 1,574 cm−1 and the I D/I G also decreases with the reduction time performed from 10 min to 6 h. These indicate that the electrochemical reduction of GO takes place within 10 min and the order of sp2 carbon atoms also increases with the prolonged electrochemical reduction time.

Raman spectra of GO and ER-GO with a reduction time of 10 min and 6 h. All of the spectra correspond to an exciting laser wavelength of 514.5 nm
TGA graphs in Fig. 7 show the weight profiles of the samples with the variation in temperature (heating rate 10°/min) under N2 flow. As for GO, weight loss of 10 wt% up to 100 °C is ascribed to the evaporation of water molecules in the sample [20]. A small amount of weight loss in this temperature region is due to the fact that ER-GO does not contain much water as earlier reported [21]. As the temperature increases from 150 to 210 °C, GO exhibits a significant weight loss of 21 wt%, indicating the evaporation of water and removal of labile oxygen functional groups [21], while the weight loss of ER-GO reduced for 10 min and 6 h coincides very well from 30 to 400 °C, with much less weight from 150 to 210 °C. After the temperature increases to 400 °C, the ER-GO lost weight more rapidly in the case of 10-min reduction than that of 6-h reduction, which suggests that a large amount of labile oxygen groups are removed simultaneously by electrochemical reduction and the degree of reduction increases with the prolonged reduction time [22].

TGA graphs of GO and ERGO reduced for different times
This result is also confirmed by XPS analysis. As shown in Fig. 8a, b, the content of O decreases and the C/O ratio increases remarkably after 10 min of electrochemical reduction. As shown in Fig. 8c, d, the C 1s XPS spectra of GO and ER-GO indicate the presence of four types of carbon bonds: C–C (284.6 eV), C–O (286.6 eV), C=O (287.6 eV), and O–C=O (289.0 eV). By comparing the XPS spectra of GO and ER-GO obtained by 4 h of reduction, it can be seen that the spectra show obvious suppression of epoxy/ether groups, indicating that most of the epoxide and hydroxyl functional groups are effectively removed. The C/O ratio of GO, ER-GO reduced for 10 min, and ER-GO reduced for 4 h is 1.5, 3.1, and 3.8, respectively, suggesting that electrochemical reduction can be effectively performed in solution by increasing the reduction time. These results obtained from XPS analysis, combined with those of XRD, FT-IR, TGA, indicate the formation of graphene through this electrochemical reduction.

XPS survey spectra of GO survey (a) and ER-GO reduced for 10 min (b) and C 1s XPS spectra of GO (c) and ER-GO reduced for 4 h (d)
The electrochemical capacitance behavior of ER-GO and CNTs is characterized by cyclic voltammetry and galvanostatic charged/discharge methods. As can be seen in Fig. 9, the ER-GO produces a much larger redox peak at about 0.3 V than that of GO, presenting a larger part of capacitance for ER-GO. This indicates that increased capacitive effects should be attributed to the reduction of oxygen groups and the GO with too many oxygen groups has very low capacitance because of its low conductivity.

Cyclic voltammetry curves of GO and ER-GO in 1 M H2SO4. The scan rate is 10 mV/s
Figure 10 shows the cyclic voltammograms (CVs) of the ER-GO at various scan rates in the range of −0.2 to 0.8 V. The appearance of one pair of redox peaks may be caused by the typical transition between quinine/hydroquinone groups of carbon materials [23, 24]. The CVs are close to be rectangular as the scan rates increase, especially at 50 mV/s, suggesting that ER-GO has an excellent capacitance behavior. This is because ER-GO could offer both sides to the electrolyte with a high surface area, leading to high specific capacitance.

Cyclic voltammetry curves of ER-GO in 1 M H2SO4 at a scan rate increasing from 5, 10, 20, to 50 mV
The specific capacitance is calculated by the following equation:
where i is the discharged current, Δt is the discharged time, and ΔV is the potential window. The charge and discharge curves of GO and ER-GO are shown in Fig. 11 where the capacitance of GO is 6.3 F g−1, and after reduction for 10 min, the capacitance of ER-GO increases to 67.7 F g−1 much higher than that of CNTs.

Charge/discharge curves of GO and ER-GO at a current density of 0.5 A g−1
The capacitance of ER-GO after reduction for 2 and 4 h increases to 148.5 and 150.5 F g−1, respectively, which are higher than the previously reported value of 117 F g−1 in H2SO4 [25]. With the time of 8 h, the capacitance increases to 158.5 F g−1, which is almost the same as that reported by Shao et al. using an electrochemical reduction method [10]. When the reduction time prolonged to 12 h, the capacitance slightly reduces to 152 F g−1. These results indicate that GO can be electrochemically reduced within 10 min and capacitance demonstrates a prominent increase within the prolonged reduction time of 8 h. After reduction for 8 h, the electrochemical reduction of GO is becoming difficult due to the fact that the remaining oxygen-containing groups are stable under electrochemical conditions and the prolonged reduction time may lead to an increase of conglomeration of ER-GO sheets by van der Waals force, resulting in the decrease of capacitance.
The electrochemical stability of the ER-GO reduced for 8 h was examined by charge/discharge cycling at a current density of 0.5 A g−1. As shown in Fig. 12, after slightly increasing capacitance during the first 500 cycles, the ER-GO reveals a stable capacitance of 154 F g−1. This result indicates that this kind of material has a long-term electrochemical stability. Furthermore, during the cycling process, the coulombic efficiency (discharge/charge) remains close to 96 %. Therefore, ER-GO is promising as an electrode material for supercapacitors, being more and more important in energy storage.

Cycle life and coulombic efficiency of ER-GO at a constant current density of 0.5 A g−1
The mechanism of the electrochemical reduction in solution can be explained as shown in Fig. 1: the dispersed GO solution is stirred rapidly using a magnetic stirring apparatus so that the GO flake is able to rapidly contact with the graphite electrolyzer and Pt plate, which serve as cathode and anode. During this process, the electrochemical reduction could be performed repeatedly. With the solution being stirred, the GO sheets would have more time to contact with the cathode and anode, resulting in the fast reduction of GO. The carboxylate and carbonyl functional groups at the edges of GO produced by the Hummers method could be reduced at the anode electrode and the possible reaction procedure could be as follows:
The first step is oxidation of carboxylate; after that, happen oxidative decarboxylation may happen. The third step is dimerization of radicals [15]. The reaction scheme above is similar to the Kolbe reaction. When in contact with the anode, GO platelets could give the electrons away, leading to the oxidation of carboxylate groups on the periphery of GO sheets. The unpaired electrons formed by the Kolbe-like loss of CO2 migrating through the GO framework could form covalent bonds after finding unpaired electrons. Radicals mostly form two-electron bonds on graphene platelets. The carboxylate group could also be reduced to hydroxy at the cathode electrode. This decarboxylation mechanism may explain the loss of carboxylates and carbonyl groups in the IR spectra, which has also been confirmed by Guo et al. [12]. Although the epoxide and hydroxyl groups are hard to be eliminated, they could also be reduced at the cathode at negative potential [12]. The mechanism of this reduction is still under discussion.
Conclusions
A novel electrochemical reduction to synthesize graphene is reported in this paper. This method opens a new mass production way for the environmentally friendly preparation of graphene. This method also presents green, time-saving, high-yield, low-cost, low-temperature features in comparison with the conventional chemical method. The capacitance of ER-GO is much higher than that of GO and carbon nanotubes. This electrochemically reduced graphene oxide is a promising material for further application in supercapacitors or electrocatalysis.
References
Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA (2004) Science 306:666–669
Zhu YW, Murali S, Cai WW, Li XS, Suk JW, Potts JR, Ruoff RS (2010) Adv Mater 22:3906–3924
Singh V, Joung D, Zhai L, Das S, Khondaker SI, Seal S (2011) Prog Mater Sci 56:1178–1271
Allen MJ, Tung VC, Kaner RB (2010) Chem Rev 110:132–145
Nagashima A, Nuka K, Satoh K, Itoh H, Ichinokawa T, Oshima C, Otani S (1993) Surf Sci 287:609–613
Berger C, Song ZM, Li TB, Li XB, Ogbazghi AY, Feng R, Dai ZT (2004) J Phys Chem B 108:19912–19916
Nethravathi C, Rajamathi M (2008) Carbon 46:1994–1998
Dreyer DR, Murali S, Zhu YW, Ruoff RS, Bielawski CW (2011) J Mater Chem 21:3443–3447
Fan XB, Peng WC, Li Y, Li XY, Wang SL, Zhang GL, Zhang FB (2008) Adv Mater 20:4490–4493
Shao YY, Wang J, Engelhard M, Wang CM, Lin YH (2010) J Mater Chem 20:743–748
Liu CB, Wang K, Luo SL, Tang YH, Chen LY (2011) Small 9:1203–1206
Guo HL, Wang XF, Qian QY, Wang FB, Xia XH (2009) ACS Nano 9:2653–2659
Liu N, Luo F, Wu HX, Liu YH, Zhang C, Chen J (2008) Adv Funct Mater 18:1518–1525
Dilimon VS, Sampath S (2011) Thin Solid Films 519:2323–2327
An SJ, Zhu YW, Lee SH, Stoller MD, Emilsson T, Park SJ, Velamakanni A, An J, Ruoff RS (2010) J Phys Chem Lett 1:1259–1263
Park S, An J, Piner RD, Jung I, Yang D, Velamakanni A, Nguyen ST, Ruoff RS (2008) Chem Mater 20:6592–6594
Xu YX, Bai H, Lu GW, Li C, Shi GQ (2008) J Am Chem Soc 130:5856–5857
Casiraghi C, Hartschuh A, Qian H, Piscanec S, Georgi C, Fasoli A, Novoselov K, Basko D, Ferrari A (2009) Nano Lett 9:1433–1441
Kudin KN, Ozbas B, Schniepp HC, Prud'homme RK, Aksay LA, Car R (2008) Nano Lett 8:36–41
Stankovich S, Dikin DA, Piner RD, Kohlhaas KA, Kleinhammes A, Jia YY, Wu Y, Nguyen ST, Ruoff RS (2007) Carbon 45:1558–1565
Lerf A, He HY, Forster M, Klinowski J (1998) J Phys Chem B 102:4477–4482
Park SJ, An J, Potts JR, Velamakanni A, Murali S, Ruoff RS (2011) Carbon 49:3019–3022
Chen Y, Zhang X, Zhang DC, Yu P, Ma YW (2011) Carbon 49:573–580
Wang DW, Li F, Zhao JP, Ren WC, Chen ZG, Tan J (2009) ACS Nano 3:1745–1753
Vivekchand SRC, Rout CS, Subrahmanyam KS, Govindaraj A, Rao CNR (2008) J Chem Sci 120:9–13
Acknowledgments
This work was supported by the Chinese Postdoctoral Foundation (2011M500910), Postdoctoral Foundation of Jiangsu Province (1201014B) and Natural Science Foundation of China (no. 21173120).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Tong, H., Zhu, J., Chen, J. et al. Electrochemical reduction of graphene oxide and its electrochemical capacitive performance. J Solid State Electrochem 17, 2857–2863 (2013). https://doi.org/10.1007/s10008-013-2195-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-013-2195-z