
Abstract
Poly(2-amino-5-(4-pyridinyl)-1,3,4-thiadiazole) (PAPT) modified glassy carbon electrode (GCE) was fabricated and used for the simultaneous determinations of dopamine (DA), uric acid (UA) and nitrite (NO2 −) in 0.1 mol L−1 phosphate buffer solution (PBS, pH 5.0) by using cyclic voltammetry and differential pulse voltammetry (DPV) techniques. The results showed that the PAPT modified GCE (PAPT/GCE) not only exhibited electrocatalytic activities towards the oxidation of DA, UA and NO2 − but also could resolve the overlapped voltammetric signals of DA, UA and NO2 − at bare GCE into three strong and well-defined oxidation peaks with enhanced current responses. The peak potential separations are 130 mV for DA–UA and 380 mV for UA–NO2 − using DPV, which are large enough for the simultaneous determinations of DA, UA and NO2 −. Under the optimal conditions, the anodic peak currents were correspondent linearly to the concentrations of DA, UA and NO2 − in the ranges of 0.95–380 μmol L−1, 2.0–1,000 μmol L−1 and 2.0–1,200 μmol L−1 for DA, UA and NO2 −, respectively. The correlation coefficients were 0.9989, 0.9970 and 0.9968, and the detection limits were 0.2, 0.35 and 0.6 μmol L−1 for DA, UA and NO2 −, respectively. In 0.1 mol L−1 PBS pH 5.0, the PAPT film exhibited good electrochemical activity, showing a surface-controlled electrode process with the apparent heterogeneous electron transfer rate constant (k s) of 25.9 s−1 and the charge–transfer coefficient (α) of 0.49, and thus displayed the features of an electrocatalyst. Due to its high sensitivity, good selectivity and stability, the modified electrode had been successfully applied to the determination of analytes in serum and urine samples.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dopamine (DA) is one of the important catecholamine neurotransmitters in the mammalian central nervous system [1, 2]. As a cholinergic drug, DA is widely applied to the treatment of circulatory collapse syndrome caused by the myocardial infarction, trauma, cardiac surgery or congestive cardiac failure. The basal DA concentration in the extracellular fluid of the central nervous system is about 0.1 μmol L−1 [3], and the abnormal levels of DA may result in some serious diseases such as parkinsonism [4, 5]. Uric acid (UA), a primary end product of purine metabolism, is another important compound present in urine and blood. The typical concentrations of UA is in millimolar range (120–450 μmol L−1) in blood and about 2 mmol L−1 in urine [6, 7], and its abnormality can lead to some diseases like gout, hyperpiesia, and Lesch–Nyhan disease [8, 9]. Nitrite is a naturally occurring form of nitrogen in aquatic systems and mainly present in the environment due to the wide use of nitrogenous chemical fertilizers. It is known that excessive nitrite in food and water can lead to many detrimental effects on human health, especially for infants and pregnant women. Thus, many techniques have been used to detect nitrite, including chemiluminescence [10], electrochemical analyses [11, 12], flame atomic absorption spectrometry [13] and spectrophotometry [14], etc. Also, it has been found that nitrogen oxide (NO) can enhance or inhibit the release of DA [15, 16] in the central nervous system, and NO can be oxidized to nitrite (NO2 −) in biological circumstance. In healthy human subjects, NO2 − can be detected at levels of 0.50–3.6 μmol L−1 in plasma, ~15 μmol L−1 in respiratory tract lining fluids, 30–210 μmol L−1 in saliva, and 0.40–60 μmol L−1 in gastric juice [17]. Therefore, the development of sensitive and selective biosensors for their simultaneous detection is highly desirable for analytical applications and diagnostic researches. However, it is nearly impossible to determine them simultaneously at bare electrodes due to the weak and overlapped oxidation waves and the electrode fouling caused by the adsorption of their oxidation products, which can lead to the poor sensitivity and selectivity [18]. To overcome these problems, various modifiers have been coated on electrode surfaces to construct the sensors for the simultaneous detections of these species, such as carbon nanotubes incorporated with the La(OH)3 nanorods [19], single-walled carbon nanohorns [20], conductive polymer [21], nanoparticles [22] and iron-doped carbon nanotubes [23]. Recently, Yuan and coworkers have fabricated the Au-nanoclusters incorporated poly(3-amino-5-mercapto-1,2,4-triazole) film [24] and the lanthanum multi-walled carbon nanotube nanocomposite film [25] modified glassy carbon electrode using electrodeposition method and used it for the simultaneous determinations of ascorbic acid, DA, UA and nitrite with satisfactory results.
Conducting polymers have attracted considerable interest in recent years because of their actual and potential applications in different fields, such as, the corrosion protection [26], electrochromic materials [27], photocatalysis [28] and sensors [29]. It is well-known that conducting polymers can be synthesized by using chemical and electrochemical techniques. The chemical polymerization technique is very useful for obtaining large amounts of polymer while the electrochemical procedure is more suitable for gaining smooth polymers on substrate surfaces and investigating the properties of polymers in situ. In addition, the electropolymerization procedure can produce thickness controllable, insoluble and uniform polymer films on the electrode surfaces. Some conventional polymers, such as, polyacetylene [30, 31], polythiophene [32, 33], polypyrrole [34, 35] and polyaniline [36, 37], have been studied extensively in the past. The synthesis and characterization of new conducting polymers can enlarge the diversity of the materials with special properties. Recently, thiazole-based conjugated polymers have attracted more and more attention because they exhibit excellent selectivity and sensitivity towards the detection of several biomolecules with stable voltammetric response [38, 39]. And also, Yang’s research group has studied the surface-enhanced Raman scattering spectrum of APD on the silver surface, and the results explored that the APT molecule with a tilted orientation anchored at the silver surface and exhibited the sound anticorrosive effect with an efficiency of 89.5 % [40].
Although several studies have been reported for the simultaneous determination of DA, UA and NO2 − [19, 23–25], the fabrication of an electrochemical sensor with high sensitivity and selectivity is still one of the paramount tasks for analytical chemists. In the present work, we found that the electropolymerized poly(2-amino-5-(4-pyridinyl)-1,3,4-thiadiazole) (PAPT) film on glassy carbon electrode (GCE) surface not only showed good electrocatalytic activities towards the oxidation of DA, UA and NO2 − but also led to the resolution of the overlapped voltammetric signals of these species at bare GCE into three strong and well-defined oxidation peaks with enhanced current responses. Thus, the selective and simultaneous determinations of DA, UA and NO2 − at the PAPT-modified GCE has been investigated in detail, and also, the modified electrode has been used to detect them in real samples.
Materials and methods
Reagents and chemicals
2-Amino-5-(4-pyridinyl)-1,3,4-thiadiazole (APT) was purchased from TCI (Japan). DA and UA were purchased from Sigma (USA). Sodium nitrite (NaNO2) was purchased from Shanghai Zhenxing Chemical Factory. Phosphate buffer solutions (PBS, 0.1 mol L−1) with various pH values were prepared using 0.1 mol L−1 Na2HPO4, 0.1 mol L−1 NaH2PO4 and 0.1 mol L−1 KCl. Twice-distilled water was used throughout the experiments.
Apparatus
All electrochemical experiments were performed with a CHI 660C electrochemical workstation (Shanghai Chenhua). The conventional three-electrode system includes a PAPT-modified GCE as working electrode, a saturated calomel reference electrode and a platinum wire auxiliary electrode.
Differential pulse voltammetry (DPV) experiments employed a scan rate of 20 mV s−1, a pulse amplitude of 25 mV, a pulse rate of 0.5 s, a pulse width of 60 ms and a quiet time of 2 s. The solutions were deoxygenated by bubbling purified nitrogen, and a nitrogen atmosphere was maintained over the solutions. All experiments were made at room temperature (≈24 °C).
Preparation of modified electrode
Glassy carbon electrode (GCE; diameter, 3.0 mm) was respectively polished with 0.3 and 0.05 μm alumina slurry, and then rinsed ultrasonically with ethanol and water for several minutes, successively.
Electropolymerization of APT on GCE was carried out by 15 successive potential sweeps between −0.2 and 1.7 V at 0.05 V s−1 in 0.1 mol L−1 H2SO4 solution containing 1 mmol L−1 APT. To remove any physisorbed, unreacted materials from the electrode surface, the electrode was rinsed with ethanol and water and sonicated for 2 min in 0.1 mol L−1 PBS pH 5.0. Then, the PAPT-modified GCE (PAPT/GCE) was obtained and stored in 0.1 mol L−1 PBS pH 5.0 at 4 °C for use.
Results and discussion
Electropolymerization of APT on GCE
Figure 1 shows the potentiodynamic polymerization (15 cycles) of 1 mmol L−1 APT in 0.1 mol L−1 H2SO4 solution in the potential window from −0.20 to 1.70 V at GCE. As can be seen, in the first cycle, APT shows a pair of oxidation and reduction peaks located at 1.34 and 0.32 V, respectively, and in the subsequent cycle, a new pair of redox waves can be observed at 0.36 and 0.32 V. The oxidation peak in the 1st cycle at 1.34 V is attributed to the oxidation of the amine to a radical cation. It is known that the oxidation of an amine group is usually observed around 0.80 V for aniline and their derivatives [41] while the presence of hetero-atoms (two N and one S atom) in the APT ring may have shifted the oxidation potential of the amine to 1.34 V, and the reduction wave at 0.32 V in the 1st cycle is due to the electro-reduction of the formed electroactive PAPT produced by the cross-reaction of the APT radical cations. And, the pair of redox waves located at 0.36/0.32 V from the 2nd potential scan is due to the oxidation and reduction of the formed PAPT produced by the cross-reaction of the APT radical cations. On the other hand, one can also see that the current response for the oxidation peak at 1.34 V in the 1st cycle decreases with the continuous scan. This is due to the rapid decrease of the amount of APT radical cations in solution, resulted by the cross-reaction/polymerization of the APT radical cations into PAPT. While the peak current responses for the redox waves at 0.36/0.32 V from the 2nd cycle increase gradually with the continuous potential scan, this indicates that the amount of PAPT on electrode surface increases gradually. These typical electrochemical behaviors not only exhibit clearly the occurrence of the electropolymerization of APT but also show that the formed polymer is electroactive and can be oxidized at a lower potential, which is necessary for being used as an electrocatalyst. The proposed mechanism for the electrochemical polymerization of the APT is illustrated in Scheme S1 in the Electronic Supplementary Material, which is supported by the results reported by Ye and coworkers [42].

CVs of GCE in 0.1 mol L−1 H2SO4 solution containing 1 mmol L−1 APT. Scan rate, 0.05 V s−1
Electrochemical behavior of the PAPT film
The cyclic voltammograms (CVs) of the PAPT film on the GCE surface deposited by 15, 50 and 100 potential sweeps in monomer-free 0.1 mol L−1 H2SO4 are shown in Fig. 2a. It can be seen from Fig. 2a that the PAPT shows a couple of redox waves located at about 0.32/0.36 V, which is due to the proton and electron addition and elimination reaction at the –NH– sites in the PAPT structure. One can also find that the oxidation and reduction peak currents increase gradually with the continuous polymerization by potential scan from 15 to 100 cycles. These results not only suggest the formation of the electroactive PAPT on electrode surface but also exhibit the gradual increasing amount of PAPT on GCE surface with the increasing potential scan. However, in the subsequent studies, we have used the 15-cycle-deposited PAPT film-modified GCE because it showed the largest peak potential separations and the highest sensitivities for the simultaneous detections of DA, UA and NO2 − in their mixture, as shown in Fig. S1 in the Electronic Supplementary Material.

a CVs of PAPT/GCE deposited by 15 (a), 50 (b) and 100 (c) potential cycles in 0.1 mol L−1 PBS pH 5.0 at 0.05 V s−1. b CVs of PAPT/GCE in 0.1 mol L−1 PBS pH 5.0 at different scan rates. Scan rates from inner to outer are 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.12, 0.14, 0.16, 0.18, 0.2, 0.24, 0.28, 0.32, 0.36, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and 1.0 V s−1, respectively. Inset: the plot of anodic peak currents vs. scan rates (top), and the variation of peak potential vs. the logarithm of scan rate (bottom)
To further investigate the electrochemical behavior of the PAPT film on the electrode surface, the PAPT/GCE was put into the APT-free 0.1 mol L−1 H2SO4 solution for potential scan at different scan rates (Fig. 2b). As can be seen, the anodic peak current increases linearly with the increasing scan rate (Fig. 2b, inset, top), which indicates that the electrode reaction is a surface-controlled process, confirming that the PAPT on electrode surface is stable. Furthermore, it can be seen from Fig. 2b that, with the increasing of scan rate, the anodic peak shifts to more positive potential while the cathodic peak shifts to more negative potential. The anodic and cathodic peak potentials are linearly dependent on the logarithm of scan rates (υ) when υ > 0.12 V s−1, this result is in agreement with Laviron’s theory [43]: a plot of E p versus log υ yields two straight lines with the slops of −2.3RT/αnF for the cathodic peak and 2.3RT/(1 − α)nF for the anodic peak (Fig. 2b, inset, bottom). So that α can be estimated as 0.49 from the slope of the straight lines based on Eq. (1). And, the average kinetic parameter k s = 25.9 s−1 can be obtained according to Eq. (2).
Electrochemical impedance spectroscopy
Electrochemical impedance spectroscopy (EIS) has been employed to investigate the impedance changes of the electrode surfaces in the modified process. EIS includes a semicircular part and a linear part; the semicircular part at high frequencies corresponds to an electron transfer limited process, and the linear part at low frequencies to a diffusion process. The EIS results for GCE and PAPT/GCE at an open circuit were shown in Fig. S2 in the Electronic Supplementary Material. To understand clearly the electrical properties of the electrode/solution interfaces, the Randles equivalent circuit (inset of Fig. S2) was chosen to fit for the obtained impedance data [44]. In the Randles circuit, it was assumed that the resistance to charge transfer (R ct) and the diffusion impedance (W) were both in parallel to the interfacial capacity (C dl). This parallel combination of R ct and C dl gives rise to a semicircle in the complex plane plot of Z″ against Z′; the semicircular diameter equals the charge–transfer resistance (R ct). This resistance exhibits the electron transfer kinetics of the redox probe at electrode interface. As shown in Fig. S2, the R ct is about 60 Ω for GCE. While after modification with PAPT film, the R ct increases dramatically to 1,000 Ω, indicating that the PAPT film hinders the charge transfer. Meanwhile, the change of R ct value in the modification process indicates that the PAPT film has been successfully coated on the GCE surface.
Electrochemical oxidation of DA, UA and NO2 −
To investigate the electrocatalytic activity of PAPT/GCE for the electrooxidation of DA, UA and NO2 −, the voltammetric responses of DA, UA and NO2 − at GCE (curve a) and PAPT/GCE (curve b) are shown in Fig. 3a, b, c, respectively.

CVs of 15 μmol L−1 DA (a), 200 μmol L−1 UA (b) and 500 μmol L−1 NO2 − (c) at GCE (a) and PAPT/GCE (b) in 0.1 mol L−1 PBS pH 5.0. Scan rate, 0.05 V s−1
Firstly, we can see that there is a pair of well-defined redox waves located at about 0.01/0.085 V on the CVs at Fig. 3(a → c); this should be ascribed to the oxidation and reduction of the electro-active PAPT on the GCE surface because this couple of redox waves always appear in the following electrochemical measurements, although there are some small changes in the redox peak potential. However, one can see that there is a big difference between this redox potential and the results shown in Fig. 2, in which the PAPT/GCE is potentially scanned only in the 0.1 mol L−1 PBS pH 5.0. The detailed mechanism that the coexisted DA, UA and NO2 − can lead to the negative shift of the redox potential of PAPT cannot be given clearly at present; however, it is rational to think that the oxidation and reduction of the coexisted DA, UA and NO2 − can accelerate the electron transfer between PAPT and electrode, and thus improve the electrochemical behavior of PAPT. This phenomenon has also been observed for the electrochemical behavior of poly(calconcarboxylic acid) (PCCA) [45], in which the redox peak potential of PCCA only in 0.05 mol L−1 H2SO4 is 0.3/0.4 V while when norepinephrine or UA coexisted, its potential shifted negatively to 0/0.10 V.
Secondly, it can be seen from Fig. 3a that DA shows a broad and weak oxidation peak at 0.31 V at the bare GCE (ΔE p = 0.14 V); however, a sharp and well-defined anodic peak appears at 0.26 V at the PAPT/GCE (ΔE p = 0.02 V). As for the current response, it has about a 4.2-fold increase in anodic peak signal at PAPT/GCE compared with that at the bare GCE. The negatively shifted oxidation potential and the improved peak current response exhibit that the PAPT-modified GCE plays an excellent catalytic effect towards the electrooxidation of DA. Figure 3b shows that at the bare GCE, UA exhibits a sluggish and much smaller CV peak response at 0.40 V while at the PAPT/GCE, UA shows a sharp and strong oxidation peak with a greatly increased peak current response, which is about a 4.2-fold increase in comparison with that at bare GCE. This indicates that the PAPT/GCE can catalyze the electrooxidation of UA. Also, from Fig. 3c, one can see that at the bare GCE, NO2 − only shows a small and broad oxidation peak at 0.94 V; however, a sharp and strong oxidation peak appears at 0.86 V at PAPT/GCE. More over, about 3.7-fold increase in peak current at PAPT/GCE can be observed in comparison with that at the bare GCE. The negatively shifted oxidation peak potential and the increased peak current response show that the PAPT/GCE exhibits an excellent catalytic effect on the electrooxidation of NO2 −.
Voltammetric separation for the anodic waves of DA and UA
It is known that the direct electrooxidation of DA and UA at bare electrodes not only requires high overpotentials but also their oxidation potentials are too closed to be distinguished. To investigate the potential application of the PAPT/GCE for the voltammetric separation of DA and UA, Fig. 4 shows the electrooxidation of 50 μmol L−1 DA and 100 μmol L−1 UA in their mixture using DPV method. Just as shown in Fig. 4, the bare GCE shows a broad and overlapped oxidation wave for the oxidation of DA and UA mixture (Fig. 4, curve a) while the presence of the PAPT film on the GCE surface can resolve the small and overlapped voltammetric response of DA and UA at the bare GCE into two sharp and well-defined oxidation peaks at 0.20 and 0.32 V (Fig. 4, curve b), corresponding to the electrooxidation of DA and UA, respectively. The separation between the two peak potentials is 0.12 V, which is large enough for the selective and simultaneous determinations of DA and UA in their mixture. Moreover, one can see that the anodic peak current responses of DA and UA at PAPT/GCE increase greatly compared with those at the bare GCE. The large peak separation degree of DA and UA anodic peaks and the greatly increased peak current responses allow us to simultaneously determine these species in their mixture with high sensitivity.

DPVs of a mixture containing 50 μmol L−1 DA and 100 μmol L−1 UA at GCE (a) and PAPT/GCE (b) in 0.1 mol L−1 PBS pH 5.0
Influence of pH
The effect of electrolyte solutions with different pH values on the electrochemical responses of DA, UA and NO2 − has been studied. Figure S3 (Electronic Supplementary Material) shows the influences of the solution pH values from 3.0 to 9.0 on the electrochemical responses of 50 μmol L−1 DA, 250 μmol L−1 UA and 500 μmol L−1 NO2 − at PAPT/GCE. It can be seen from Fig. S3 that the peak currents of DA, UA and NO2 − increase with the increasing pH value from 3.0 to 5.0 and reach a maximum at pH 5.0 then they decrease when the pH value increases gradually. Considering the sensitivity in sensing DA, UA and NO2 −, 0.1 mol L−1 PBS pH 5.0 was selected for detecting these species. On the other hand, one can also see that all the anodic peak potentials for the electrooxidation of DA, UA and NO2 − shift negatively with increasing pH; this indicates that protons take part in their electrode reaction processes.
Selective determination of DA and UA
To investigate the practicality of PAPT/GCE for the selective determination of DA and UA in their mixture, Fig. 5a, b illustrates the DPV curves recorded at the PAPT/GCE in solutions containing various concentrations of DA in the presence of 150 μmol L−1 UA and different contents of UA in the presence of 20 μmol L−1 DA, respectively. Figure 5a shows that the peak current response for the oxidation of DA increases linearly with the increase of DA concentration while the peak current for UA oxidation keeps nearly unchanged. This indicates that the presence of UA does not interfere with the response of DA oxidation thus the proposed method can be used for the selective determination of DA in the presence of UA. Similarly, it can be seen from Fig. 5b that with the increasing UA concentration, the oxidation current of UA exhibits a linear increase while the peak response of DA keeps almost stable. This indicates that the presence of DA does not interfere with the response of UA oxidation, and thus, the proposed method can be used for the selective determination of UA in the presence of DA.

a DPVs of DA with different concentrations at PAPT/GCE in the presence of 150 μmol L−1 UA in 0.1 mol L−1 PBS pH 5.0. DA concentrations (a→j): 0.5, 3, 30, 55, 70, 95, 120, 140, 160 and 200 μmol L−1. b DPVs of different concentrations of UA at PAPT/GCE in the presence of 20 μmol L−1 DA in 0.1 μmol L−1 PBS pH 5.0. UA concentrations (a→j): 20, 80, 150, 220, 280, 350, 420, 500, 600 and 700 μmol L−1
Simultaneous determination of DA and UA
To investigate the possibility of PAPT/GCE for the simultaneous determinations of DA and UA, the current responses of these species by simultaneously changing the concentrations of DA and UA in a mixture have been measured (Fig. 6). As can be seen, the peak currents for the electrooxidation of DA and UA increase proportionally with their concentrations. The linear relationships between the oxidation peak currents i p,a (microampere) and the concentrations c (micromole per liter) of DA and UA are: i p,a = 0.102 + 0.158c DA (r = 0.9998) and i p,a = 1.016 + 0.023c UA (r = 0.9997), respectively. The linear ranges for the simultaneous determinations of DA and UA are 0.95–74 and 2.5–600 μmol L−1, respectively. The detection limits for detecting DA and UA are 0.15 and 0.3 μmol L−1 (S/N = 3), respectively.

DPVs of DA and UA mixtures at PAPT/GCE in 0.1 mol L−1 PBS pH 5.0. DA concentrations (a→j): 0.95, 2.3, 10, 18, 27, 34, 47, 61, 70 and 74 μmol L−1, UA concentrations (a→j): 2.5, 35, 80, 115, 170, 240, 330, 450, 560 and 600 μmol L−1
Voltammetric separation for the anodic waves of DA, UA and NO2 −
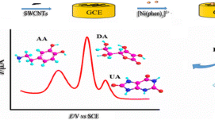
Figure 7 shows the electrooxidation of 350 μmol L−1 DA, 350 μmol L−1 UA and 150 μmol L−1 NO2 − in their mixture by using DPV method. It is obvious that the bare GCE (Fig. 7, curve a) only gives a broad and overlapped anodic peak for DA and UA, and an inconspicuous anodic peak for NO2 −. While at the PAPT/GCE, three sharp and well-defined oxidation peaks at 0.26, 0.39 and 0.77 V (Fig. 7, curve b) are observed, corresponding to the electrooxidation of DA, UA and NO2 −, respectively. Moreover, the peak current responses of DA, UA and NO2 − are notably improved compared with those at the bare GCE. The large peak separations of the anodic peaks for these three compounds and the increased current responses allow us to simultaneously determine these species in their mixture with high sensitivity.

DPVs of a mixture containing 350 μmol L−1 DA, 350 μmol L−1 UA and 150 μmol L−1 NO2 − at GCE (a) and PAPT/GCE (b) in 0.1 mol L−1 PBS pH 5.0
Selective determination of DA, UA and NO2 −
The selective determination of DA, UA and NO2 − at PAPT/GCE in their mixture has been investigated when the concentration of one species changed, and those of the other two species keep constant; the results are shown in Fig. 8. Figure 8a shows that the peak current of DA increases with the increasing DA concentration while the peak current of UA and NO2 − keeps nearly unchanged. Similarly, Fig. 8b, c shows that the peak current responses of UA and NO2 − increase with the increasing of their concentrations while the peak currents of the other two compounds keep nearly stable. This indicates that the presence of the other two species does not interfere with the response of the other species, therefore, the PAPT/GCE can be employed for the selective determination of DA, UA and NO2 − in the presence of the other two species.

a DPVs of DA with different concentrations at PAPT/GCE in the presence of 200 μmol L−1 UA and 80 μmol L−1 NO2 − in 0.1 mol L−1 PBS pH 5.0. DA concentrations (a→j): 0.5, 10, 40, 100, 160, 220, 280, 340, 400 and 480 μmol L−1. b DPVs of UA with different concentrations at PAPT/GCE in the presence of 150 μmol L−1 DA and 80 μmol L−1 NO2 − in 0.1 μmol L−1 PBS pH 5.0. UA concentrations (a→j): 30, 100, 150, 330, 520, 700, 850, 1,000, 1,100 and 1,250 μmol L−1. c DPVs of NO2 − with different concentrations at PAPT/GCE in the presence of 10 μmol L−1 DA and 100 μmol L−1 UA in 0.1 μmol L−1 PBS pH 5.0. NO2 − concentrations (a→j): 2.0, 30, 80, 130, 300, 460, 580, 700, 1,150 and 1,600 μmol L−1
Simultaneous determination of DA, UA and NO2 −
Figure 9 shows the DPV results of different concentrations of DA, UA and NO2 − in a mixture at PAPT/GCE in 0.1 mol L−1 PBS pH 5.0. It can be seen from Fig. 9 that the peak current responses of DA, UA and NO2 − increase with the rising of their concentrations, and the peak currents are linearly proportional to the concentrations of DA, UA and NO2 − in the ranges of 0.95–380, 2.0–1,000 and 2.0–1,200 μmol L−1, respectively. The linear relationships between the peak currents i p,a (microampere) and concentrations c (micromole per liter) of DA, UA and NO2 − are: i p,a = 0.678 + 0.0261c DA (r = 0.9989), i p,a = 0.0965 + 0.0059c UA (r = 0.9970) and i p,a = 0.181 + 0.0076c NO2− (r = 0.9968), respectively. The detection limits for detecting DA, UA and NO2 − are 0.2, 0.35, and 0.6 μmol L−1 (S/N = 3), respectively. Compared with the other related studies on the simultaneous detections of DA, UA and NO2 −, as shown in Table S1 in the Electronic Supplementary Material, the present method exhibits large peak potential separations, high sensitivities, and wide linear ranges.

DPVs of DA, UA and NO2 − mixture at PAPT/GCE in 0.1 mol L−1 PBS pH 5.0. DA concentrations (a→i): 0.95, 50, 90, 150, 190, 220, 260, 300 and 380 μmol L−1; UA concentrations (a→i): 2.0, 50, 100, 150, 250, 400, 550, 700 and 1,000 μmol L−1; NO2 − concentrations (a→i): 2.0, 100, 200, 320, 450, 600, 750, 950 and 1,200 μmol L−1
Interferences, stability and reproducibility
For investigating the anti-interference ability of the PAPT/GCE, several co-existing substances were selected. No significant interference for the detection of DA (15 μmol L−1), UA (30 μmol L−1) and NO2 − (45 μmol L−1) was observed from the following compounds (micromole per liter): NaCl (500), KCl (500), CaCl2 (500), MgSO4 (500), ZnCl2 (500), glucose (350), NH4 + (300), l-cysteine (300) and ascorbic acid (250). The stability of the PAPT film-modified GCE was also tested. When the modified electrode was kept in 0.1 mol L−1 PBS pH 5.0 at room temperature after voltammetric measurements, the current signals kept nearly stable in the first 2 days and decreased about 3.1 %, 2.5 % and 2.4 % of its initial responses for DA, UA and NO2 − after another 2 weeks, indicating the good stability of the modified electrode. To study the reproducibility of the PAPT/GCE, repetitive measurements had been carried out for simultaneous detections of 200 μmol L−1 DA, 400 μmol L−1 UA and 500 μmol L−1 NO2 − in their mixture. The results of 15 successive measurements showed relative standard deviations of 2.6 %, 3.2 % and 2.9 % for DA, UA and NO2 −, respectively. The precision at renewed surface of the PAPT/GCE was also investigated, which showed the relative standard deviations of 2.3 %, 2.7 % and 3.3 % for the determinations of 200 μmol L−1 DA, 400 μmol L−1 UA and 500 μmol L−1 NO2 −, respectively. These results indicated the high selectivity, good stability and reproducibility of the PAPT/GCE.
Sample analysis
Human urine and serum samples were selected as real samples for analysis using the standard adding method. All samples were diluted (100 times for urine and 20 times for serum) with 0.1 mol L−1 PBS pH 5.0. In order to ascertain the correctness of the results, the diluted samples mentioned above were spiked with certain amounts of DA, UA and NO2 − and then were detected. The results were presented in Tables 1 and 2, respectively. The recovery rates of the samples ranged between 96.4 % and 104.5 %, indicating that the proposed method could be effectively used for the determination of DA, UA and NO2 − in real samples.
Conclusions
In this work, PAPT film-modified GCE has been fabricated by using electrochemical oxidation of APT at GCE. The PAPT/GCE not only can improve the electrochemical behaviors of DA, UA and NO2 − with negatively shifted oxidation overpotentials and enhance the peak current responses, showing the electrocatalytic activities towards the electrooxidation of DA, UA and NO2 −, but it also can resolve the overlapped oxidation peaks of these species into three sharp and well-defined anodic waves with large peak separations. Thus, the modified electrode can be used for the simultaneous determinations of DA, UA and NO2 − with good sensitivity, selectivity and high electrocatalytic activity. The proposed method has been applied for detecting DA, UA and NO2 − in real samples with satisfactory results.
References
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. II Patterns of loss of dopamine-containing neurons in Parkinson's disease Brain 122:1437–1448
Heinz A, Przuntek H, Winterer G, Pietzcker A (1995) Clinical aspects and follow-up of dopamine-induced psychoses in continuous dopaminergic therapy and their implications for the dopamine hypothesis of schizophrenic symptoms. Nervenarzt 66:662–669
Ciszewski A, Milczarek G (1999) Polyeugenol-modified platinum electrode for selective detection of dopamine in the presence of ascorbic acid. Anal Chem 71:1055–1061
Martin C (1998) The Parkinson’s puzzle, new developments in our understanding of Parkinson’s disease have generated a number of promising new treatments for this disabling condition. Chem Br 34:40–42
Wightman RM, May LJ, Michael AC (1988) Detection of dopamine dynamics in the brain. Anal Chem 60:769A–779A
Manjunatha H, Nagaraju DH, Suresh GS, Venkatesha TV (2009) Detection of uric acid in the presence of dopamine and high concentration of ascorbic acid using PDDA modified graphite electrode. Electroanalysis 21:2198–2206
Behera S, Retna Raj C (2007) Mercaptoethylpyrazine promoted electrochemistry of redox protein and amperometric biosensing of uric acid. Biosens Bioelectron 23:556–561
Zen JM, Jou JJ, Ilangovan G (1998) Selective voltammetric method for uric acid detection using preanodized Nafion-coated glassy carbon electrodes. Analyst 123:1345–1350
Popa E, Kubota Y, Tryk DA, Fujishima A (2000) Selective voltammetric amperometric detection of uric acid with oxidized diamond film electrodes. Anal Chem 72:1724–1727
Kanda Y, Taira M (2003) Flow-injection analysis method for the determination of nitrite and nitrate in natural water samples using a chemiluminescence NOx monitor. Anal Sci 19:695–699
Biagiotti V, Valentini F, Tamburri E, Terranova ML, Moscone D, Palleschi G (2007) Synthesis and characterization of polymeric films and nanotubule nets used to assemble selective sensors for nitrite detection in drinking water. Sens Actuat B 122:236–242
Liu P, Hu J (2004) Nitrite reduction in acidic solution at a GC/Eastman-AQ-Os(bpy)32+-PVP composite modified electrode. J Electroanal Chem 566:423–432
Noroozifar M, Khorasani-Motlagh M, Taheri A, Homayoonfard M (2007) Application of manganese (IV) dioxide microcolumn for determination and speciation of nitrite and nitrate using a flow injection analysis–flame atomic absorption spectrometry system. Talanta 71:359–364
Al-Okab RA, Sayed AA (2007) Novel reactions for simple and sensitive spectrophotometric determination of nitrite. Talanta 72:1239–1247
Guevara-Guzmam R, Emson PC, Kendrick KMJ (1994) Modulation of in vivo striatal transmitter release by nitric oxide and cyclic GMP. J Neurochem 62:807–810
Zhang Y, Yuan R, Chai YQ, Li WJ, Zhong X, Zhong HA (2011) Simultaneous voltammetric determination for DA, AA and NO2 – based on graphenen/poly-cyclodextrin/MWCNTs nanocomposite platform. Biosens Bioelectron 26:3977–3980
Tahboub YR (2008) Determination of nitrite and nitrate in cell culture media by reversed-phase high-performance liquid chromatography with simultaneous UV–VIS and fluorescence detection. Jordan J Chem 3:69–75
Atta NF, El-Kady MF, Galal A (2010) Simultaneous determination of catecholamines, uric acid and ascorbic acid at physiological levels using poly(N-methylpyrrole)/Pd-nanoclusters sensor. Anal Biochem 400:78–88
Zhang Y, Yuan R, Chai YQ, Zhong X, Zhong HA (2012) Carbon nanotubes incorporated with sol–gel derived La(OH)3 nanorods as platform to simultaneously determine ascorbic acid, dopamine, uric acid and nitrite. Colloids Surf B Biointer 100:185–189
Li YY, Du J, Yang JD, Liu D, Lu XQ (2012) Electrocatalytic detection of dopamine in the presence of ascorbic acid and uric acid using single-walled carbon nanotubes modified electrode. Colloids Surf B Biointer 97:32–36
Mohammad HM, Taher Y, Ahmad NG (2012) A nickel hexacyanoferrate and poly(1-naphthol) hybrid film modified electrode used in the selective electroanalysis of dopamine. Electrochim Acta 59:321–328
Tian XQ, Cheng CM, Yuan HY, Du J, Xiao D, Xie SP, Choi Martin MF (2012) Simultaneous determination of L-ascorbic acid, dopamine and uric acid with gold nanoparticles-β-cyclodextrin-graphene-modified electrode by aquare wave voltammetry. Talanta 93:79–85
Wang C, Yuan R, Chai YQ, Chen SH, Zhang Y, Hu FX, Zhang MH (2012) Non-covalent iron(III)-porphyrin functionalized muli-walled carbon nanotubes for the simultaneous determination of ascorbic acid, dopamine, uric acid and nitrite. Electrochim Acta 62:109–115
Wang C, Yuan R, Chai YQ, Zhang Y, Hu FX, Zhang MH (2011) Au-nanoclusters incorporated 3-amino-5-mercapto-1,2,4-triazole film modified electrode for the simultaneous determination of ascorbic acid, dopamine, uric acid and nitrite. Biosens Bioelectron 30:315–319
Zhang W, Yuan R, Chai YQ, Zhang Y, Chen SH (2012) A simple strategy based on lanthanum–multiwalled carbon nanotube nanocomposites for simultaneous determination of ascorbic acid, dopamine, uric acid and nitrite. Sens Actuat B 166–167:601–607
Solmaz R (2011) Eletrochemical synthesis of poly-2-aminothiazole on mild steel and its corrosion inhibiton performance. Prog Org Coat 70:122–126
Zhang C, Hua C, Wang GH, Ouyang M, Ma CN (2010) A novel multichromic copolymer via electrochemical copolymerization of (S)-1, 1’-binaphthyl-2, 2’-diyl bis(N-(6-hexanoic acid-1-yl)pyrrole) and 3, 4-ethylenedioxythiophene. Electrochim Acta 55:4103–4111
Cristante VM, Jorge SMA, Valente JPS, Saeki MJ, Florentino AO, Padilha PM (2007) TiO2 films organofunctionalized with 2-aminothiazole ligand and adsorbed Pd(II) ions applied in the photocatalytic degradation of phenol in an aqueous medium. Thin Solid Films 515:5334–5340
Zhang L, Lang QH, Shi ZG (2010) Electrochemical synthesis of three-dimensional polyaniline network on 3-aminobenzenesulfonic acid functionalized glassy garbon electrode and it application. Am J Anal Chem 1:102–112
Abdalla Araujo A, Del Nero J, Laks B (1999) Study of copolymer composed by polyacetylene and polyazine. Synth Met 101:365–366
Kijima M, Ohmura K, Shirakawa H (1999) Electrochemical synthesis of free-standing polyacetylene film with copper catalyst. Synth Met 101:58
del Valle A, Ma GM, Díaz FR, Armijo F, Del Río R (2009) Electrosynthesis of polythiophene nanowires via mesoporous silica thin film templates. Electrochem Commun 11:2117–2120
Fabre B (2001) Trifluoroborate-substituted polythiophene as a novel conjugated polymer promising for sensing and solid-state reactions. Electrochem Commun 3:549–552
Fan LZ, Maier J (2006) High-performance polypyrrole eletrode materials for redox supercapacitors. Electrochem Commun 8:937–940
Makhloufi L, Hammache H, Saidani B (2000) Electrocatalytic reduction of proton on polypyrrole coatings onto aluminium modified by the electrochemical cementation process. Electrochem Commun 2:552–556
Zhang L, Jiang X, Niu L, Dong SJ (2006) Syntheses of fully sulfonated polyaniline nano-networks and its application to the direct electrochemistry of cytochrome c. Biosens Bioelectron 21:1107–1115
Zhang L, Zhang J, Zhang CH (2009) Electrochemical synthesis of polyaniline nano-network on α-alianine functionalized glassy carbon electrode and its application for the direct electrochemistry of horse heart cytochrome c. Biosens Bioelectron 24:2085–2090
Revin SB, John SA (2011) Electropolymerization of 3-amino-5-mercapto-1, 2, 4-triazole on glassy carbon electrode and its electrocatalytic activity towards uric acid. Electrochim Acta 56:8934–8940
Xue Y, Sheng ZH, Zhao H, Wu ZJ, Li XJ, He YJ, Yuan Z (2012) Electrochemical synthesis and characterization of a novel thiazole-based copolymer and its application in biosensor. Electrochim Acta 59:256–263
Zhang LJ, Wen Y, Pan YC, Yang HF (2011) 2-Amino-5-(4-pyridinyl)-1,3,4-thiadiazole film at the silver surface: Observation by Raman spectroscopy and electrochemical methods. Appl Surf Sci 257:6347–6352
Genies EM, Lapkowski M (1988) Polyaniline films. Electrochemical redox mechanisms Synth Met 24:61–68
Ye SY, Tian SW, Wang SJ, Wang JM (1988) Electrochemical polymerization and characterization of 4-vinylpyridine. J Xiamen Univ 27:181–185
Laviron E (1979) General expression of the liner potential sweep voltammogram in the case of diffusionless electrochemical systems. J Electroanal Chem 101:19–28
Finklea HO, Snider DA, Fedyk J, Sabatani E, Gafni Y, Rubinstein I (1993) Characterization of octadecanethiol-coated gold electrodes as microarray electrodes by cyclic voltammetry and ac impedance spectroscopy. Langmuir 9:3660–3667
Liu AL, Zhang SB, Chen W, Lin XH, Xia XH (2008) Simultaneous votammetric determination of norepinephrine, ascorbic acid and uric acid on polycalconcarboxylic aid modified glassy carbon electrode. Biosens Bioelectron 23:1488–1495
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 567 kb)
Rights and permissions
About this article
Cite this article
Zhang, L., Wang, L. Poly(2-amino-5-(4-pyridinyl)-1, 3, 4-thiadiazole) film modified electrode for the simultaneous determinations of dopamine, uric acid and nitrite. J Solid State Electrochem 17, 691–700 (2013). https://doi.org/10.1007/s10008-012-1904-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-012-1904-3